
Constipation is a bowel dysfunction that makes bowel movements infrequent or hard to pass. The stool is often hard and dry. Other symptoms may include abdominal pain, bloating, and feeling as if one has not completely passed the bowel movement. Complications from constipation may include hemorrhoids, anal fissure or fecal impaction. The normal frequency of bowel movements in adults is between three per day and three per week. Babies often have three to four bowel movements per day while young children typically have two to three per day.

A rectal prolapse occurs when walls of the rectum have prolapsed to such a degree that they protrude out of the anus and are visible outside the body. However, most researchers agree that there are 3 to 5 different types of rectal prolapse, depending on whether the prolapsed section is visible externally, and whether the full or only partial thickness of the rectal wall is involved.

Diverticulosis is the condition of having multiple pouches (diverticula) in the colon that are not inflamed. These are outpockets of the colonic mucosa and submucosa through weaknesses of muscle layers in the colon wall. Diverticula do not cause symptoms in most people. Diverticular disease occurs when diverticula become clinically inflamed, a condition known as diverticulitis.
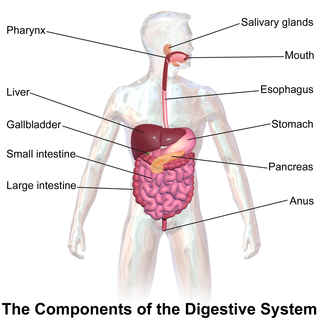
Gastrointestinal diseases refer to diseases involving the gastrointestinal tract, namely the esophagus, stomach, small intestine, large intestine and rectum, and the accessory organs of digestion, the liver, gallbladder, and pancreas.

A volvulus is when a loop of intestine twists around itself and the mesentery that supports it, resulting in a bowel obstruction. Symptoms include abdominal pain, abdominal bloating, vomiting, constipation, and bloody stool. Onset of symptoms may be rapid or more gradual. The mesentery may become so tightly twisted that blood flow to part of the intestine is cut off, resulting in ischemic bowel. In this situation there may be fever or significant pain when the abdomen is touched.
A fecal impaction or an impacted bowel is a solid, immobile bulk of feces that can develop in the rectum as a result of chronic constipation. Fecal impaction is a common result of neurogenic bowel dysfunction and causes immense discomfort and pain. Its treatment includes laxatives, enemas, and pulsed irrigation evacuation (PIE) as well as digital removal. It is not a condition that resolves without direct treatment.
Intestinal malrotation is a congenital anomaly of rotation of the midgut. It occurs during the first trimester as the fetal gut undergoes a complex series of growth and development. Malrotation can lead to a dangerous complication called volvulus, in which cases emergency surgery is indicated. Malrotation can refer to a spectrum of abnormal intestinal positioning, often including:
Intestinal neuronal dysplasia (IND) is an inherited disease of the intestine that affects one in 3000 children and adults. The intestine uses peristalsis to push its contents toward the anus; people with IND have a problem with the motor neurons that lead to the intestine, inhibiting this process and thus preventing digestion.

Megacolon is an abnormal dilation of the colon. This leads to hypertrophy of the colon. The dilation is often accompanied by a paralysis of the peristaltic movements of the bowel. In more extreme cases, the feces consolidate into hard masses inside the colon, called fecalomas, which can require surgery to be removed.

Intestinal pseudo-obstruction (IPO) is a clinical syndrome caused by severe impairment in the ability of the intestines to push food through. It is characterized by the signs and symptoms of intestinal obstruction without any lesion in the intestinal lumen. Clinical features mimic those seen with mechanical intestinal obstructions and can include abdominal pain, nausea, abdominal distension, vomiting, dysphagia and constipation depending upon the part of the gastrointestinal tract involved.
Stercoral ulcer is an ulcer of the colon due to pressure and irritation resulting from severe, prolonged constipation due to a large bowel obstruction, damage to the autonomic nervous system, or stercoral colitis. It is most commonly located in the sigmoid colon and rectum. Prolonged constipation leads to production of fecaliths, leading to possible progression into a fecaloma. These hard lumps irritate the rectum and lead to the formation of these ulcers. It results in fresh bleeding per rectum. These ulcers may be seen on imaging, such as a CT scan but are more commonly identified using endoscopy, usually a colonoscopy. Treatment modalities can include both surgical and non-surgical techniques.
A lower anterior resection, formally known as anterior resection of the rectum and colon and anterior excision of the rectum or simply anterior resection, is a common surgery for rectal cancer and occasionally is performed to remove a diseased or ruptured portion of the intestine in cases of diverticulitis. It is commonly abbreviated as LAR.
Solitary rectal ulcer syndrome or SRUS is a chronic, benign disorder of the rectal mucosa. It commonly occurs with varying degrees of rectal prolapse. The condition is thought to be caused by different factors, such as long term constipation, straining during defecation, and dyssynergic defecation. Treatment is by normalization of bowel habits, biofeedback, and other conservative measures. In more severe cases various surgical procedures may be indicated. The condition is relatively rare, affecting approximately 1 in 100,000 people per year. It affects mainly adults aged 30–50. Females are affected slightly more often than males. The disorder can be confused clinically with rectal cancer or other conditions such as inflammatory bowel disease, even when a biopsy is done.
Anismus or dyssynergic defecation is the failure of normal relaxation of pelvic floor muscles during attempted defecation. It can occur in both children and adults, and in both men and women. It can be caused by physical defects or it can occur for other reasons or unknown reasons. Anismus that has a behavioral cause could be viewed as having similarities with parcopresis, or psychogenic fecal retention.
Obstructed defecation syndrome is a major cause of functional constipation, of which it is considered a subtype. It is characterized by difficult and/or incomplete emptying of the rectum with or without an actual reduction in the number of bowel movements per week. Normal definitions of functional constipation include infrequent bowel movements and hard stools. In contrast, ODS may occur with frequent bowel movements and even with soft stools, and the colonic transit time may be normal, but delayed in the rectum and sigmoid colon.
Constipation in children refers to the medical condition of constipation in children. It is a functional gastrointestinal disorder.

Neurogenic bowel dysfunction (NBD) is the inability to control defecation due to a deterioration of or injury to the nervous system, resulting in faecal incontinence or constipation. It is common in people with spinal cord injury (SCI), multiple sclerosis (MS) or spina bifida.

Orvar Swenson was a Swedish-born American pediatric surgeon. He discovered the cause of Hirschsprung's disease and in 1948, with Alexander Bill, performed the first pull-through operation in a child with megacolon, which then became a treatment for the disease.
A pull-through procedure is the definitive operation for Hirschsprung disease, involving the removal of the abnormal segment of bowel that has no nerves, pulling through the normal bowel and connecting it to the anus. Several types of pull-through procedures exist including the Soave, Swenson and Duhamel. It can be performed using an open or minimally invasive approach.
Waardenburg syndrome type 4A is an extremely rare congenital disorder caused by a mutation in an endothelin receptor gene. It results in common Waardenburg syndrome symptoms such as abnormal hair and skin pigmentation and heterochromia, but also present with symptoms of Hirschsprung's disease. Symptoms include abdominal pain and bowel obstruction. Waardenburg syndrome type 4A is the rarest among the types, appearing only once in about every 1,000,000 individuals. There have only been a total of 50 cases reported in total as of 2016.

