Related Research Articles

Bioinformatics is an interdisciplinary field that develops methods and software tools for understanding biological data, in particular when the data sets are large and complex. As an interdisciplinary field of science, bioinformatics combines biology, computer science, information engineering, mathematics and statistics to analyze and interpret the biological data. Bioinformatics has been used for in silico analyses of biological queries using mathematical and statistical techniques.

Genomics is an interdisciplinary field of biology focusing on the structure, function, evolution, mapping, and editing of genomes. A genome is an organism's complete set of DNA, including all of its genes. In contrast to genetics, which refers to the study of individual genes and their roles in inheritance, genomics aims at the collective characterization and quantification of all of an organism's genes, their interrelations and influence on the organism. Genes may direct the production of proteins with the assistance of enzymes and messenger molecules. In turn, proteins make up body structures such as organs and tissues as well as control chemical reactions and carry signals between cells. Genomics also involves the sequencing and analysis of genomes through uses of high throughput DNA sequencing and bioinformatics to assemble and analyze the function and structure of entire genomes. Advances in genomics have triggered a revolution in discovery-based research and systems biology to facilitate understanding of even the most complex biological systems such as the brain.
Computational biology involves the development and application of data-analytical and theoretical methods, mathematical modelling and computational simulation techniques to the study of biological, ecological, behavioural, and social systems. The field is broadly defined and includes foundations in biology, applied mathematics, statistics, biochemistry, chemistry, biophysics, molecular biology, genetics, genomics, computer science, ecology, and evolution.

Eugene Viktorovich Koonin is a Russian-American biologist and Senior Investigator at the National Center for Biotechnology Information (NCBI). He is a recognised expert in the field of evolutionary and computational biology.

Comparative genomics is a field of biological research in which the genomic features of different organisms are compared. The genomic features may include the DNA sequence, genes, gene order, regulatory sequences, and other genomic structural landmarks. In this branch of genomics, whole or large parts of genomes resulting from genome projects are compared to study basic biological similarities and differences as well as evolutionary relationships between organisms. The major principle of comparative genomics is that common features of two organisms will often be encoded within the DNA that is evolutionarily conserved between them. Therefore, comparative genomic approaches start with making some form of alignment of genome sequences and looking for orthologous sequences in the aligned genomes and checking to what extent those sequences are conserved. Based on these, genome and molecular evolution are inferred and this may in turn be put in the context of, for example, phenotypic evolution or population genetics.
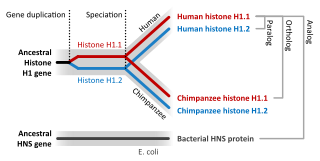
Sequence homology is the biological homology between DNA, RNA, or protein sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry because of three phenomena: either a speciation event (orthologs), or a duplication event (paralogs), or else a horizontal gene transfer event (xenologs).
The Wellcome Sanger Institute, previously known as The Sanger Centre and Wellcome Trust Sanger Institute, is a non-profit British genomics and genetics research institute, primarily funded by the Wellcome Trust.
Computational genomics refers to the use of computational and statistical analysis to decipher biology from genome sequences and related data, including both DNA and RNA sequence as well as other "post-genomic" data. These, in combination with computational and statistical approaches to understanding the function of the genes and statistical association analysis, this field is also often referred to as Computational and Statistical Genetics/genomics. As such, computational genomics may be regarded as a subset of bioinformatics and computational biology, but with a focus on using whole genomes to understand the principles of how the DNA of a species controls its biology at the molecular level and beyond. With the current abundance of massive biological datasets, computational studies have become one of the most important means to biological discovery.

In evolutionary biology, conserved sequences are identical or similar sequences in nucleic acids or proteins across species, or within a genome, or between donor and receptor taxa. Conservation indicates that a sequence has been maintained by natural selection.

Human accelerated regions (HARs), first described in August 2006, are a set of 49 segments of the human genome that are conserved throughout vertebrate evolution but are strikingly different in humans. They are named according to their degree of difference between humans and chimpanzees. Found by scanning through genomic databases of multiple species, some of these highly mutated areas may contribute to human-specific traits. Others may represent loss of functional mutations, possibly due to the action of biased gene conversion rather than adaptive evolution.

David Haussler is an American bioinformatician known for his work leading the team that assembled the first human genome sequence in the race to complete the Human Genome Project and subsequently for comparative genome analysis that deepens understanding the molecular function and evolution of the genome.

Richard Michael Durbin, FRS, born 30 December 1960, is a British computational biologist. He is currently an Associate Faculty member at the Wellcome Trust Sanger Institute and Professor of Genetics at the University of Cambridge. Previously, he was Senior Group Leader at the Wellcome Trust Sanger Institute for over 20 years and an Honorary Professor of Computational genomics at the University of Cambridge.
An ultra-conserved element (UCE) is a region of DNA that is identical in at least two different species. One of the first studies of UCEs showed that certain human DNA sequences of length 200 nucleotides or greater were entirely conserved in human, rats, and mice. Despite often being noncoding DNA, some ultra-conserved elements have been found to be transcriptionally active, giving non-coding RNA molecules.
hCONDELs refer to regions of deletions within the human genome containing sequences that are highly conserved among closely related relatives. Almost all of these deletions fall within regions that perform non-coding functions. These represent a new class of regulatory sequences and may have played an important role in the development of specific traits and behavior that distinguish closely related organisms from each other.
James Andrew Cuff, is a British biophysicist. Cuff has held leadership positions at Harvard University, the Broad Institute, The Wellcome Trust Sanger Institute and the European Bioinformatics Institute.

Alfonso Valencia is a Spanish biologist, ICREA Professor, current director of the Life Sciences department at Barcelona Supercomputing Center. and of Spanish National Bioinformatics Institute (INB-ISCIII). From 2015-2018, he was President of the International Society for Computational Biology. His research is focused on the study of biomedical systems with computational biology and bioinformatics approaches.
Mathieu Daniel Blanchette is a computational biologist and Associate Professor in the School of Computer Science at McGill University. His research focuses on developing new algorithms for the detection of functional regions in DNA sequences.
Single nucleotide polymorphism annotation is the process of predicting the effect or function of an individual SNP using SNP annotation tools. In SNP annotation the biological information is extracted, collected and displayed in a clear form amenable to query. SNP functional annotation is typically performed based on the available information on nucleic acid and protein sequences.
Christos A. Ouzounis is a computational biologist and a director of research at the CERTH in Thessaloniki.
Katherine Snowden Pollard is a Professor at the Gladstone Institute of data science and biotechnology at the University of California, San Francisco (UCSF). She was awarded Fellowship of the International Society for Computational Biology in 2020 for outstanding contributions to computational biology and bioinformatics.
References
- 1 2 Adam C. Siepel publications indexed by Google Scholar
- ↑ Adam C. Siepel's publications indexed by the Scopus bibliographic database. (subscription required)
- ↑ Brian Couger, M.; Pipes, L.; Squina, F.; Prade, R.; Siepel, A.; Palermo, R.; Katze, M. G.; Mason, C. E.; Blood, P. D. (2014). "Enabling large-scale next-generation sequence assembly with Blacklight". Concurrency and Computation: Practice and Experience. 26 (13): 2157–2166. doi:10.1002/cpe.3231. PMC 4185199 . PMID 25294974.
- ↑ ENCODE Project Consortium; Birney E; Stamatoyannopoulos JA; Dutta A; Guigó R; Gingeras TR; Margulies EH; Weng Z; Snyder M; Dermitzakis ET; et al. (2007). "Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project". Nature. 447 (7146): 799–816. Bibcode:2007Natur.447..799B. doi:10.1038/nature05874. PMC 2212820 . PMID 17571346.
- ↑ Adam Siepel's CV.
- ↑ Siepel, A. C.; Halpern, A. L.; MacKen, C; Korber, B. T. (1995). "A computer program designed to screen rapidly for HIV type 1 intersubtype recombinant sequences". AIDS Research and Human Retroviruses. 11 (11): 1413–6. doi:10.1089/aid.1995.11.1413. PMID 8573400.
- ↑ Siepel, A.; Farmer, A.; Tolopko, A.; Zhuang, M.; Mendes, P.; Beavis, W.; Sobral, B. (2001). "ISYS: A decentralized, component-based approach to the integration of heterogeneous bioinformatics resources". Bioinformatics. 17 (1): 83–94. doi: 10.1093/bioinformatics/17.1.83 . PMID 11222265.
- ↑ Siepel, A. C. (2003). "An algorithm to enumerate sorting reversals for signed permutations" (PDF). Journal of Computational Biology. 10 (3–4): 575–97. CiteSeerX 10.1.1.114.8797 . doi:10.1089/10665270360688200. PMID 12935346.
- ↑ Siepel, A.; Bejerano, G; Pedersen, J. S.; Hinrichs, A. S.; Hou, M; Rosenbloom, K; Clawson, H; Spieth, J; Hillier, L. W.; Richards, S; Weinstock, G. M.; Wilson, R. K.; Gibbs, R. A.; Kent, W. J.; Miller, W; Haussler, D (2005). "Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes". Genome Research. 15 (8): 1034–50. doi:10.1101/gr.3715005. PMC 1182216 . PMID 16024819.
- ↑ Kosiol, C.; Vinař, T. Š.; Da Fonseca, R. R.; Hubisz, M. J.; Bustamante, C. D.; Nielsen, R.; Siepel, A. (2008). "Patterns of Positive Selection in Six Mammalian Genomes". PLOS Genetics. 4 (8): e1000144. doi:10.1371/journal.pgen.1000144. PMC 2483296 . PMID 18670650.
- ↑ Pollard, K. S.; Hubisz, M. J.; Rosenbloom, K. R.; Siepel, A. (2009). "Detection of nonneutral substitution rates on mammalian phylogenies". Genome Research. 20 (1): 110–21. doi:10.1101/gr.097857.109. PMC 2798823 . PMID 19858363.
- ↑ Gronau, I.; Hubisz, M. J.; Gulko, B.; Danko, C. G.; Siepel, A. (2011). "Bayesian inference of ancient human demography from individual genome sequences". Nature Genetics. 43 (10): 1031–4. doi:10.1038/ng.937. PMC 3245873 . PMID 21926973.
- ↑ Arbiza, L.; Gronau, I.; Aksoy, B. A.; Hubisz, M. J.; Gulko, B.; Keinan, A.; Siepel, A. (2013). "Genome-wide inference of natural selection on human transcription factor binding sites". Nature Genetics. 45 (7): 723–729. doi:10.1038/ng.2658. PMC 3932982 . PMID 23749186.
- ↑ Gulko, B.; Hubisz, M. J.; Gronau, I.; Siepel, A. (2015). "A method for calculating probabilities of fitness consequences for point mutations across the human genome". Nature Genetics. 47 (3): 276–283. doi:10.1038/ng.3196. PMC 4342276 . PMID 25599402.
- 1 2 Guggenheim profile. Archived April 18, 2012, at the Wayback Machine
| General | |
|---|---|
| Scientific databases | |