Related Research Articles

A myelodysplastic syndrome (MDS) is one of a group of cancers in which immature blood cells in the bone marrow do not mature, and as a result, do not develop into healthy blood cells. Early on, no symptoms typically are seen. Later, symptoms may include fatigue, shortness of breath, bleeding disorders, anemia, or frequent infections. Some types may develop into acute myeloid leukemia.
Aplastic anemia (AA) is a severe hematologic condition in which the body fails to make blood cells in sufficient numbers. Aplastic anemia is associated with cancer and various cancer syndromes. Blood cells are produced in the bone marrow by stem cells that reside there. Aplastic anemia causes a deficiency of all blood cell types: red blood cells, white blood cells, and platelets.

Fanconi anemia (FA) is a rare, AR, genetic disease resulting in impaired response to DNA damage in the FA/BRCA pathway. Although it is a very rare disorder, study of this and other bone marrow failure syndromes has improved scientific understanding of the mechanisms of normal bone marrow function and development of cancer. Among those affected, the majority develop cancer, most often acute myelogenous leukemia (AML), MDS, and liver tumors. 90% develop aplastic anemia by age 40. About 60–75% have congenital defects, commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and developmental disabilities. Around 75% have some form of endocrine problem, with varying degrees of severity. 60% of FA is FANC-A, 16q24.3, which has later onset bone marrow failure.

Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, acquired, life-threatening disease of the blood characterized by destruction of red blood cells by the complement system, a part of the body's innate immune system. This destructive process occurs due to deficiency of the red blood cell surface protein DAF, which normally inhibits such immune reactions. Since the complement cascade attacks the red blood cells within the blood vessels of the circulatory system, the red blood cell destruction (hemolysis) is considered an intravascular hemolytic anemia. There is ongoing research into other key features of the disease, such as the high incidence of venous blood clot formation. Research suggests that PNH thrombosis is caused by both the absence of GPI-anchored complement regulatory proteins on PNH platelets and the excessive consumption of nitric oxide (NO).
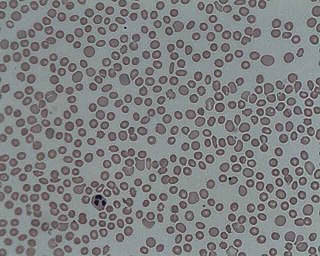
In hematology, thrombocytopenia is a condition characterized by abnormally low levels of platelets in the blood. Low levels of platelets in turn may lead to prolonged or excessive bleeding. It is the most common coagulation disorder among intensive care patients and is seen in a fifth of medical patients and a third of surgical patients.
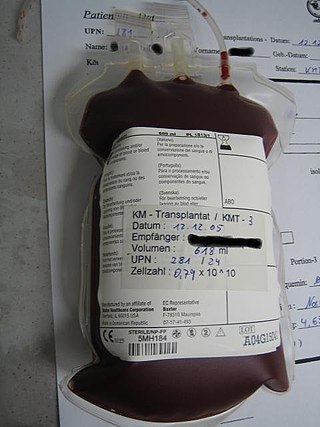
Hematopoietic stem-cell transplantation (HSCT) is the transplantation of multipotent hematopoietic stem cells, usually derived from bone marrow, peripheral blood, or umbilical cord blood in order to replicate inside of a patient and to produce additional normal blood cells. It may be autologous, allogeneic or syngeneic.
Cytopenia is a reduction in the number of mature blood cells. It can have many causes, and commonly occurs in people with cancer being treated with radiation therapy or chemotherapy.
Pancytopenia is a medical condition in which there is significant reduction in the number of almost all blood cells.
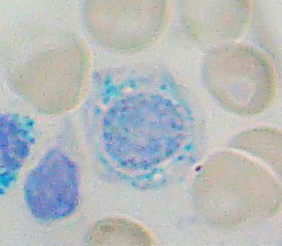
Sideroblastic anemia, or sideroachrestic anemia, is a form of anemia in which the bone marrow produces ringed sideroblasts rather than healthy red blood cells (erythrocytes). In sideroblastic anemia, the body has iron available but cannot incorporate it into hemoglobin, which red blood cells need in order to transport oxygen efficiently. The disorder may be caused either by a genetic disorder or indirectly as part of myelodysplastic syndrome, which can develop into hematological malignancies.

Dyskeratosis congenita (DKC), also known as Zinsser-Engman-Cole syndrome, is a rare progressive congenital disorder with a highly variable phenotype. The entity was classically defined by the triad of abnormal skin pigmentation, nail dystrophy, and leukoplakia of the oral mucosa, and MDS/AML, but these components do not always occur. DKC is characterized by short telomeres. Some of the manifestations resemble premature ageing and cognitive impairment can be a feature. The disease initially mainly affects the skin, but a major consequence is progressive bone marrow failure which occurs in over 80%, causing early mortality.
Mean platelet volume (MPV) is a machine-calculated measurement of the average size of platelets found in blood and is typically included in blood tests as part of the CBC. Since the average platelet size is larger when the body is producing increased numbers of platelets, the MPV test results can be used to make inferences about platelet production in bone marrow or platelet destruction problems.
Aplasia is a birth defect where an organ or tissue is wholly or largely absent. It is caused by a defect in a developmental process.
Reticulocytopenia is the medical term for an abnormal decrease in circulating red blood cell precursors (reticulocytes) that can lead to anemia due to resulting low red blood cell (erythrocyte) production. Reticulocytopenia may be an isolated finding or it may not be associated with abnormalities in other hematopoietic cell lineages such as those that produce white blood cells (leukocytes) or platelets (thrombocytes), a decrease in all three of these lineages is referred to as pancytopenia.
The National Marrow Donor Program (NMDP) is a nonprofit organization founded in 1986 and based in Minneapolis, Minnesota, that operates the Be The Match Registry of volunteer hematopoietic cell donors and umbilical cord blood units in the United States.
Pearson syndrome is a mitochondrial disease characterized by sideroblastic anemia and exocrine pancreas dysfunction. Other clinical features are failure to thrive, pancreatic fibrosis with insulin-dependent diabetes and exocrine pancreatic deficiency, muscle and neurologic impairment, and, frequently, early death. It is usually fatal in infancy. The few patients who survive into adulthood often develop symptoms of Kearns–Sayre syndrome. It is caused by a deletion in mitochondrial DNA. Pearson syndrome is very rare, less than a hundred cases have been reported in medical literature worldwide.
Acute myelomonocytic leukemia (AMML) is a form of acute myeloid leukemia that involves a proliferation of CFU-GM myeloblasts and monoblasts. AMML occurs with a rapid increase amount in white blood cell count and is defined by more than 20% of myeloblast in the bone marrow. It is classified under "M4" in the French-American-British classification (FAB). It is classified under "AML, not otherwise classified" in the WHO classification.

Congenital amegakaryocytic thrombocytopenia (CAMT) is a rare autosomal recessive bone marrow failure syndrome characterized by severe thrombocytopenia, which can progress to aplastic anemia and leukemia. CAMT usually manifests as thrombocytopenia in the initial month of life or in the fetal phase. Typically CAMPT presents with petechiae, cerebral bleeds, recurrent rectal bleeding, or pulmonary hemorrhage.
Hoyeraal–Hreidasson syndrome is a very rare multisystem X-linked recessive disorder characterized by excessively short telomeres and is considered a severe form of dyskeratosis congenita. Being an X-linked disorder, Hoyeraal–Hreidasson syndrome primarily affects males. Patients typically present in early childhood with cerebellar hypoplasia, immunodeficiency, progressive bone marrow failure, and intrauterine growth restriction. The primary cause of death in Hoyeraal–Hreidasson syndrome is bone marrow failure, but mortality from cancer and pulmonary fibrosis is also significant.
Refractory cytopenia of childhood is a subgroup of myelodysplastic syndrome (MDS), having been added to the World Health Organization classification in 2008. Before then, RCC cases were classified as childhood aplastic anemia. RCC is the most common form of MDS in children and adolescents, accounting for approximately half of all MDS cases.
GATA2 deficiency is a grouping of several disorders caused by common defect, namely, familial or sporadic inactivating mutations in one of the two parental GATA2 genes. Being the gene haploinsufficient, mutations that cause a reduction in the cellular levels of the gene's product, GATA2, are autosomal dominant. The GATA2 protein is a transcription factor critical for the embryonic development, maintenance, and functionality of blood-forming, lymphatic-forming, and other tissue-forming stem cells. In consequence of these mutations, cellular levels of GATA2 are deficient and individuals develop over time hematological, immunological, lymphatic, or other presentations that may begin as apparently benign abnormalities but commonly progress to severe organ failure, opportunistic infections, virus infection-induced cancers, the myelodysplastic syndrome, and/or leukemia. GATA2 deficiency is a life-threatening and precancerous condition.
References
- ↑ "Bone Marrow Failure In Children - What You Need to Know". www.drugs.com. Archived from the original on 2018-06-25. Retrieved 2017-01-31.
- ↑ Kitchen, Rose. "Signs & Symptoms of Bone Marrow Failure". eHow Health. Demand Media, Inc (2011): 1-4.
{{cite web}}: Missing or empty|url=(help) - ↑ "Bone Marrow Failure In Children". Thomson Reuters (2011): 1-5. Archived from the original on 25 June 2018. Retrieved 7 Nov 2011.
- 1 2 Besa, Emmanuel C. "Bone Marrow Failure". Medscape Reference: Drugs, Diseases & Procedures. WebMD, LLC, (2011): 1-5.
{{cite web}}: Missing or empty|url=(help) - ↑ Nakao, Shinji (7 January 2023). "Diagnosis of immune pathophysiology in patients with bone marrow failure". International Journal of Hematology. doi:10.1007/s12185-022-03519-1.
- ↑ Voit, R.A., Sankaran, V.G. MECOM Deficiency: from Bone Marrow Failure to Impaired B-Cell Development. J Clin Immunol 43, 1052–1066 (2023). https://doi.org/10.1007/s10875-023-01545-0
- 1 2 "Blood and bone marrow donation definition". Mayo Clinic. Retrieved 6 December 2011.
- ↑ Blanche, Alter (January 2018). "Cancer in the National Cancer Institute Inherited Bone Marrow Failure Syndrome Cohort After Fifteen Years of Follow-Up". Hemaematologica - Via MEDLINE (EBSCO).
- 1 2 3 4 Moore, Christine (January 2019). "Bone Marrow Failure". StatPearls.
- ↑ Ashraf, Malouf (May 2018). "Comparison of a therapeutic-only versus prophylactic platelet transfusion policy for people with congenital or acquired bone marrow failure disorders (Review)". Cochrane Database of Systematic Reviews.
- 1 2 3 4 Nagalla, Srikanth (19 July 2021). "Bone Marrow Failure". Medscape.
- 1 2 3 4 MDS Foundation. "What is MDS?". Myelodysplastic Syndromes Foundation.
- 1 2 Leguit, Roos J; Jan G. van den Tweel (2010). "The pathology of bone marrow failure" (PDF). Histopathology. 57 (5): 655–670. doi:10.1111/j.1365-2559.2010.03612.x. PMID 20727024. S2CID 1807526.
- ↑ "Aplastic Anemia". Health and Wellness Magazine. 12 December 2010.