
Haematopoiesis is the formation of blood cellular components. All cellular blood components are derived from haematopoietic stem cells. In a healthy adult human, roughly ten billion to a hundred billion new blood cells are produced per day, in order to maintain steady state levels in the peripheral circulation.

A myelodysplastic syndrome (MDS) is one of a group of cancers in which immature blood cells in the bone marrow do not mature, and as a result, do not develop into healthy blood cells. Early on, no symptoms typically are seen. Later, symptoms may include fatigue, shortness of breath, bleeding disorders, anemia, or frequent infections. Some types may develop into acute myeloid leukemia.

Fanconi anemia (FA) is a rare, AR, genetic disease resulting in impaired response to DNA damage in the FA/BRCA pathway. Although it is a very rare disorder, study of this and other bone marrow failure syndromes has improved scientific understanding of the mechanisms of normal bone marrow function and development of cancer. Among those affected, the majority develop cancer, most often acute myelogenous leukemia (AML), MDS, and liver tumors. 90% develop aplastic anemia by age 40. About 60–75% have congenital defects, commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and developmental disabilities. Around 75% have some form of endocrine problem, with varying degrees of severity. 60% of FA is FANC-A, 16q24.3, which has later onset bone marrow failure.
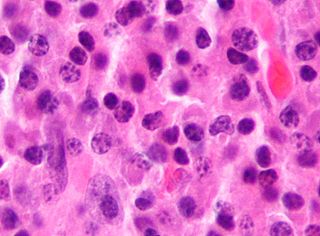
Tumors of the hematopoietic and lymphoid tissues or tumours of the haematopoietic and lymphoid tissues are tumors that affect the blood, bone marrow, lymph, and lymphatic system. Because these tissues are all intimately connected through both the circulatory system and the immune system, a disease affecting one will often affect the others as well, making aplasia, myeloproliferation and lymphoproliferation closely related and often overlapping problems. While uncommon in solid tumors, chromosomal translocations are a common cause of these diseases. This commonly leads to a different approach in diagnosis and treatment of hematological malignancies. Hematological malignancies are malignant neoplasms ("cancer"), and they are generally treated by specialists in hematology and/or oncology. In some centers "hematology/oncology" is a single subspecialty of internal medicine while in others they are considered separate divisions. Not all hematological disorders are malignant ("cancerous"); these other blood conditions may also be managed by a hematologist.
Primary myelofibrosis (PMF) is a rare bone marrow blood cancer. It is classified by the World Health Organization (WHO) as a type of myeloproliferative neoplasm, a group of cancers in which there is activation and growth of mutated cells in the bone marrow. This is most often associated with a somatic mutation in the JAK2, CALR, or MPL genes. In PMF, the bony aspects of bone marrow are remodeled in a process called osteosclerosis; in addition, fibroblast secrete collagen and reticulin proteins that are collectively referred to as (fibrosis). These two pathological processes compromise the normal function of bone marrow resulting in decreased production of blood cells such as erythrocytes, granulocytes and megakaryocytes, the latter cells responsible for the production of platelets.

Acute myeloid leukemia (AML) is a cancer of the myeloid line of blood cells, characterized by the rapid growth of abnormal cells that build up in the bone marrow and blood and interfere with normal blood cell production. Symptoms may include feeling tired, shortness of breath, easy bruising and bleeding, and increased risk of infection. Occasionally, spread may occur to the brain, skin, or gums. As an acute leukemia, AML progresses rapidly, and is typically fatal within weeks or months if left untreated.

Chronic myelomonocytic leukemia (CMML) is a type of leukemia, which are cancers of the blood-forming cells of the bone marrow. In adults, blood cells are formed in the bone marrow, by a process that is known as haematopoiesis. In CMML, there are increased numbers of monocytes and immature blood cells (blasts) in the peripheral blood and bone marrow, as well as abnormal looking cells (dysplasia) in at least one type of blood cell.

Cluster of differentiation antigen 135 (CD135) also known as fms like tyrosine kinase 3, receptor-type tyrosine-protein kinase FLT3, or fetal liver kinase-2 (Flk2) is a protein that in humans is encoded by the FLT3 gene. FLT3 is a cytokine receptor which belongs to the receptor tyrosine kinase class III. CD135 is the receptor for the cytokine Flt3 ligand (FLT3L).

ETV6 protein is a transcription factor that in humans is encoded by the ETV6 gene. The ETV6 protein regulates the development and growth of diverse cell types, particularly those of hematological tissues. However, its gene, ETV6 frequently suffers various mutations that lead to an array of potentially lethal cancers, i.e., ETV6 is a clinically significant proto-oncogene in that it can fuse with other genes to drive the development and/or progression of certain cancers. However, ETV6 is also an anti-oncogene or tumor suppressor gene in that mutations in it that encode for a truncated and therefore inactive protein are also associated with certain types of cancers.
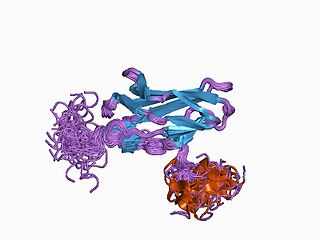
Runt-related transcription factor 1 (RUNX1) also known as acute myeloid leukemia 1 protein (AML1) or core-binding factor subunit alpha-2 (CBFA2) is a protein that in humans is encoded by the RUNX1 gene.

Homeobox protein Hox-A9 is a protein that in humans is encoded by the HOXA9 gene.

GATA2 or GATA-binding factor 2 is a transcription factor, i.e. a nuclear protein which regulates the expression of genes. It regulates many genes that are critical for the embryonic development, self-renewal, maintenance, and functionality of blood-forming, lympathic system-forming, and other tissue-forming stem cells. GATA2 is encoded by the GATA2 gene, a gene which often suffers germline and somatic mutations which lead to a wide range of familial and sporadic diseases, respectively. The gene and its product are targets for the treatment of these diseases.

PHD finger protein 6 is a protein that in humans is encoded by the PHF6 gene.

Acute megakaryoblastic leukemia (AMKL) is life-threatening leukemia in which malignant megakaryoblasts proliferate abnormally and injure various tissues. Megakaryoblasts are the most immature precursor cells in a platelet-forming lineage; they mature to promegakaryocytes and, ultimately, megakaryocytes which cells shed membrane-enclosed particles, i.e. platelets, into the circulation. Platelets are critical for the normal clotting of blood. While malignant megakaryoblasts usually are the predominant proliferating and tissue-damaging cells, their similarly malignant descendants, promegakaryocytes and megakaryocytes, are variable contributors to the malignancy.

Tet methylcytosine dioxygenase 2 (TET2) is a human gene. It resides at chromosome 4q24, in a region showing recurrent microdeletions and copy-neutral loss of heterozygosity (CN-LOH) in patients with diverse myeloid malignancies.
Graft-versus-tumor effect (GvT) appears after allogeneic hematopoietic stem cell transplantation (HSCT). The graft contains donor T cells that can be beneficial for the recipient by eliminating residual malignant cells. GvT might develop after recognizing tumor-specific or recipient-specific alloantigens. It could lead to remission or immune control of hematologic malignancies. This effect applies in myeloma and lymphoid leukemias, lymphoma, multiple myeloma and possibly breast cancer. It is closely linked with graft-versus-host disease (GvHD), as the underlying principle of alloimmunity is the same. CD4+CD25+ regulatory T cells (Treg) can be used to suppress GvHD without loss of beneficial GvT effect. The biology of GvT response is still not fully understood but it is probable that the reaction with polymorphic minor histocompatibility antigens expressed either specifically on hematopoietic cells or more widely on a number of tissue cells or tumor-associated antigens is involved. This response is mediated largely by cytotoxic T lymphocytes (CTL) but it can be employed by natural killers as separate effectors, particularly in T-cell-depleted HLA-haploidentical HSCT.

Musashi-2, also known as Musashi RNA binding protein 2, is a protein that in humans is encoded by the MSI2 gene. Like its homologue musashi-1 (MSI1), it is an RNA-binding protein involved in stemness.
Clonal hypereosinophilia, also termed primary hypereosinophilia or clonal eosinophilia, is a grouping of hematological disorders all of which are characterized by the development and growth of a pre-malignant or malignant population of eosinophils, a type of white blood cell that occupies the bone marrow, blood, and other tissues. This population consists of a clone of eosinophils, i.e. a group of genetically identical eosinophils derived from a sufficiently mutated ancestor cell.
GATA2 deficiency is a grouping of several disorders caused by common defect, namely, familial or sporadic inactivating mutations in one of the two parental GATA2 genes. Being the gene haploinsufficient, mutations that cause a reduction in the cellular levels of the gene's product, GATA2, are autosomal dominant. The GATA2 protein is a transcription factor critical for the embryonic development, maintenance, and functionality of blood-forming, lymphatic-forming, and other tissue-forming stem cells. In consequence of these mutations, cellular levels of GATA2 are deficient and individuals develop over time hematological, immunological, lymphatic, or other presentations that may begin as apparently benign abnormalities but commonly progress to severe organ failure, opportunistic infections, virus infection-induced cancers, the myelodysplastic syndrome, and/or leukemia. GATA2 deficiency is a life-threatening and precancerous condition.
Christopher Hourigan is a physician-scientist known for work on measurable residual disease in acute myeloid leukemia.
