
A DNA polymerase is a member of a family of enzymes that catalyze the synthesis of DNA molecules from nucleoside triphosphates, the molecular precursors of DNA. These enzymes are essential for DNA replication and usually work in groups to create two identical DNA duplexes from a single original DNA duplex. During this process, DNA polymerase "reads" the existing DNA strands to create two new strands that match the existing ones. These enzymes catalyze the chemical reaction

DNA repair is a collection of processes by which a cell identifies and corrects damage to the DNA molecules that encodes its genome. In human cells, both normal metabolic activities and environmental factors such as radiation can cause DNA damage, resulting in tens of thousands of individual molecular lesions per cell per day. Many of these lesions cause structural damage to the DNA molecule and can alter or eliminate the cell's ability to transcribe the gene that the affected DNA encodes. Other lesions induce potentially harmful mutations in the cell's genome, which affect the survival of its daughter cells after it undergoes mitosis. As a consequence, the DNA repair process is constantly active as it responds to damage in the DNA structure. When normal repair processes fail, and when cellular apoptosis does not occur, irreparable DNA damage may occur, including double-strand breaks and DNA crosslinkages. This can eventually lead to malignant tumors, or cancer as per the two-hit hypothesis.
RecQ helicase is a family of helicase enzymes initially found in Escherichia coli that has been shown to be important in genome maintenance. They function through catalyzing the reaction ATP + H2O → ADP + P and thus driving the unwinding of paired DNA and translocating in the 3' to 5' direction. These enzymes can also drive the reaction NTP + H2O → NDP + P to drive the unwinding of either DNA or RNA.

Hematopoietic stem cells (HSCs) are the stem cells that give rise to other blood cells. This process is called haematopoiesis. In vertebrates, the very first definitive HSCs arise from the ventral endothelial wall of the embryonic aorta within the (midgestational) aorta-gonad-mesonephros region, through a process known as endothelial-to-hematopoietic transition. In adults, haematopoiesis occurs in the red bone marrow, in the core of most bones. The red bone marrow is derived from the layer of the embryo called the mesoderm.

Non-homologous end joining (NHEJ) is a pathway that repairs double-strand breaks in DNA. It is called "non-homologous" because the break ends are directly ligated without the need for a homologous template, in contrast to homology directed repair (HDR), which requires a homologous sequence to guide repair. NHEJ is active in both non-dividing and proliferating cells, while HDR is not readily accessible in non-dividing cells. The term "non-homologous end joining" was coined in 1996 by Moore and Haber.

Terminal deoxynucleotidyl transferase (TdT), also known as DNA nucleotidylexotransferase (DNTT) or terminal transferase, is a specialized DNA polymerase expressed in immature, pre-B, pre-T lymphoid cells, and acute lymphoblastic leukemia/lymphoma cells. TdT adds N-nucleotides to the V, D, and J exons of the TCR and BCR genes during antibody gene recombination, enabling the phenomenon of junctional diversity. In humans, terminal transferase is encoded by the DNTT gene. As a member of the X family of DNA polymerase enzymes, it works in conjunction with polymerase λ and polymerase μ, both of which belong to the same X family of polymerase enzymes. The diversity introduced by TdT has played an important role in the evolution of the vertebrate immune system, significantly increasing the variety of antigen receptors that a cell is equipped with to fight pathogens. Studies using TdT knockout mice have found drastic reductions (10-fold) in T-cell receptor (TCR) diversity compared with that of normal, or wild-type, systems. The greater diversity of TCRs that an organism is equipped with leads to greater resistance to infection. Although TdT was one of the first DNA polymerases identified in mammals in 1960, it remains one of the least understood of all DNA polymerases. In 2016–18, TdT was discovered to demonstrate in trans template dependant behaviour in addition to its more broadly known template independent behaviour

Werner syndrome ATP-dependent helicase, also known as DNA helicase, RecQ-like type 3, is an enzyme that in humans is encoded by the WRN gene. WRN is a member of the RecQ Helicase family. Helicase enzymes generally unwind and separate double-stranded DNA. These activities are necessary before DNA can be copied in preparation for cell division. Helicase enzymes are also critical for making a blueprint of a gene for protein production, a process called transcription. Further evidence suggests that Werner protein plays a critical role in repairing DNA. Overall, this protein helps maintain the structure and integrity of a person's DNA.
V(D)J recombination is the mechanism of somatic recombination that occurs only in developing lymphocytes during the early stages of T and B cell maturation. It results in the highly diverse repertoire of antibodies/immunoglobulins and T cell receptors (TCRs) found in B cells and T cells, respectively. The process is a defining feature of the adaptive immune system.

Ku is a dimeric protein complex that binds to DNA double-strand break ends and is required for the non-homologous end joining (NHEJ) pathway of DNA repair. Ku is evolutionarily conserved from bacteria to humans. The ancestral bacterial Ku is a homodimer. Eukaryotic Ku is a heterodimer of two polypeptides, Ku70 (XRCC6) and Ku80 (XRCC5), so named because the molecular weight of the human Ku proteins is around 70 kDa and 80 kDa. The two Ku subunits form a basket-shaped structure that threads onto the DNA end. Once bound, Ku can slide down the DNA strand, allowing more Ku molecules to thread onto the end. In higher eukaryotes, Ku forms a complex with the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) to form the full DNA-dependent protein kinase, DNA-PK. Ku is thought to function as a molecular scaffold to which other proteins involved in NHEJ can bind, orienting the double-strand break for ligation.

DNA repair protein XRCC4 also known as X-ray repair cross-complementing protein 4 or XRCC4 is a protein that in humans is encoded by the XRCC4 gene. In addition to humans, the XRCC4 protein is also expressed in many other metazoans, fungi and in plants. The X-ray repair cross-complementing protein 4 is one of several core proteins involved in the non-homologous end joining (NHEJ) pathway to repair DNA double strand breaks (DSBs).

DNA-dependent protein kinase, catalytic subunit, also known as DNA-PKcs, is an enzyme that in humans is encoded by the gene designated as PRKDC or XRCC7. DNA-PKcs belongs to the phosphatidylinositol 3-kinase-related kinase protein family. The DNA-Pkcs protein is a serine/threonine protein kinase consisting of a single polypeptide chain of 4,128 amino acids.

DNA ligase 4 is an enzyme that in humans is encoded by the LIG4 gene.
DNA polymerase lambda, also known as Pol λ, is an enzyme found in all eukaryotes. In humans, it is encoded by the POLL gene.

DNA polymerase theta is an enzyme that in humans is encoded by the POLQ gene. This polymerase plays a key role in one of the three major double strand break repair pathways: theta-mediated end joining (TMEJ). Most double-strand breaks are repaired by non-homologous end joining (NHEJ) or homology directed repair (HDR). However, in some contexts, NHEJ and HR are insufficient and TMEJ is the only solution to repair the break. TMEJ is often described as alternative NHEJ, but differs in that it lacks a requirement for the Ku heterodimer, and it can only act on resected DNA ends. Following annealing of short regions on the DNA overhangs, DNA polymerase theta catalyzes template-dependent DNA synthesis across the broken ends, stabilizing the paired structure.
Non-homologous end-joining factor 1 (NHEJ1), also known as Cernunnos or XRCC4-like factor (XLF), is a protein that in humans is encoded by the NHEJ1 gene. XLF was originally discovered as the protein mutated in five patients with growth retardation, microcephaly, and immunodeficiency. The protein is required for the non-homologous end joining (NHEJ) pathway of DNA repair. Patients with XLF mutations also have immunodeficiency due to a defect in V(D)J recombination, which uses NHEJ to generate diversity in the antibody repertoire of the immune system. XLF interacts with DNA ligase IV and XRCC4 and is thought to be involved in the end-bridging or ligation steps of NHEJ. The yeast homolog of XLF is Nej1.

NAD-dependent deacetylase sirtuin 7 is an enzyme that in humans is encoded by the SIRT7 gene. SIRT7 is member of the mammalian sirtuin family of proteins, which are homologs to the yeast Sir2 protein.
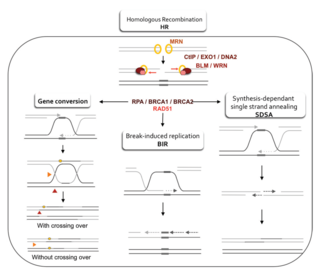
Homology-directed repair (HDR) is a mechanism in cells to repair double-strand DNA lesions. The most common form of HDR is homologous recombination. The HDR mechanism can only be used by the cell when there is a homologous piece of DNA present in the nucleus, mostly in G2 and S phase of the cell cycle. Other examples of homology-directed repair include single-strand annealing and breakage-induced replication. When the homologous DNA is absent, another process called non-homologous end joining (NHEJ) takes place instead.
Microhomology-mediated end joining (MMEJ), also known as alternative nonhomologous end-joining (Alt-NHEJ) is one of the pathways for repairing double-strand breaks in DNA. As reviewed by McVey and Lee, the foremost distinguishing property of MMEJ is the use of microhomologous sequences during the alignment of broken ends before joining, thereby resulting in deletions flanking the original break. MMEJ is frequently associated with chromosome abnormalities such as deletions, translocations, inversions and other complex rearrangements.

DNA end resection, also called 5′–3′ degradation, is a biochemical process where the blunt end of a section of double-stranded DNA (dsDNA) is modified by cutting away some nucleotides from the 5' end to produce a 3' single-stranded sequence. The presence of a section of single-stranded DNA (ssDNA) allows the broken end of the DNA to line up accurately with a matching sequence, so that it can be accurately repaired.

A double-strand break repair model refers to the various models of pathways that cells undertake to repair double strand-breaks (DSB). DSB repair is an important cellular process, as the accumulation of unrepaired DSB could lead to chromosomal rearrangements, tumorigenesis or even cell death. In human cells, there are two main DSB repair mechanisms: Homologous recombination (HR) and non-homologous end joining (NHEJ). HR relies on undamaged template DNA as reference to repair the DSB, resulting in the restoration of the original sequence. NHEJ modifies and ligates the damaged ends regardless of homology. In terms of DSB repair pathway choice, most mammalian cells appear to favor NHEJ rather than HR. This is because the employment of HR may lead to gene deletion or amplification in cells which contains repetitive sequences. In terms of repair models in the cell cycle, HR is only possible during the S and G2 phases, while NHEJ can occur throughout whole process. These repair pathways are all regulated by the overarching DNA damage response mechanism. Besides HR and NHEJ, there are also other repair models which exists in cells. Some are categorized under HR, such as synthesis-dependent strain annealing, break-induced replication, and single-strand annealing; while others are an entirely alternate repair model, namely, the pathway microhomology-mediated end joining (MMEJ).







