Chromatin is a complex of DNA, RNA, and protein found in eukaryotic cells. Its primary function is packaging very long DNA molecules into a more compact, denser shape, which prevents the strands from becoming tangled and plays important roles in reinforcing the DNA during cell division, preventing DNA damage, and regulating gene expression and DNA replication. During mitosis and meiosis, chromatin facilitates proper segregation of the chromosomes in anaphase; the characteristic shapes of chromosomes visible during this stage are the result of DNA being coiled into highly condensed networks of chromatin.

The Encyclopedia of DNA Elements (ENCODE) is a public research project which aims to identify functional elements in the human genome.
An active chromatin sequence (ACS) is a region of DNA in a eukaryotic chromosome in which histone modifications such as acetylation lead to exposure of the DNA sequence thus allowing binding of transcription factors and transcription to take place. Active chromatin may also be called euchromatin. ACSs may occur in non-expressed gene regions which are assumed to be "poised" for transcription. The sequence once exposed often contains a promoter to began transcription. At this site acetylation or methylation can take place causing a conformational change to the chromatin. At the active chromatin sequence site deacetylation can caused the gene to be repressed if not being expressed.
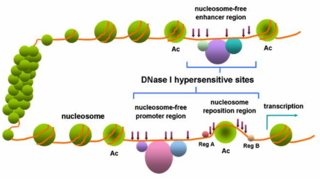
In genetics a hypersensitive site is a short region of chromatin and is detected by its super sensitivity to cleavage by DNase I and other various nucleases. In a hypersensitive site, the nucleosomal structure is less compacted, increasing the availability of the DNA to binding by proteins, such as transcription factors and DNase I. These sites account for many inherited tendencies.
ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global binding sites precisely for any protein of interest. Previously, ChIP-on-chip was the most common technique utilized to study these protein–DNA relations.
DamID is a molecular biology protocol used to map the binding sites of DNA- and chromatin-binding proteins in eukaryotes. DamID identifies binding sites by expressing the proposed DNA-binding protein as a fusion protein with DNA methyltransferase. Binding of the protein of interest to DNA localizes the methyltransferase in the region of the binding site. Adenosine methylation does not occur naturally in eukaryotes and therefore adenine methylation in any region can be concluded to have been caused by the fusion protein, implying the region is located near a binding site. DamID is an alternate method to ChIP-on-chip or ChIP-seq.

Tiling arrays are a subtype of microarray chips. Like traditional microarrays, they function by hybridizing labeled DNA or RNA target molecules to probes fixed onto a solid surface.
Epigenomics is the study of the complete set of epigenetic modifications on the genetic material of a cell, known as the epigenome. The field is analogous to genomics and proteomics, which are the study of the genome and proteome of a cell. Epigenetic modifications are reversible modifications on a cell's DNA or histones that affect gene expression without altering the DNA sequence. Epigenomic maintenance is a continuous process and plays an important role in stability of eukaryotic genomes by taking part in crucial biological mechanisms like DNA repair. Plant flavones are said to be inhibiting epigenomic marks that cause cancers. Two of the most characterized epigenetic modifications are DNA methylation and histone modification. Epigenetic modifications play an important role in gene expression and regulation, and are involved in numerous cellular processes such as in differentiation/development and tumorigenesis. The study of epigenetics on a global level has been made possible only recently through the adaptation of genomic high-throughput assays.
DNA binding sites are a type of binding site found in DNA where other molecules may bind. DNA binding sites are distinct from other binding sites in that (1) they are part of a DNA sequence and (2) they are bound by DNA-binding proteins. DNA binding sites are often associated with specialized proteins known as transcription factors, and are thus linked to transcriptional regulation. The sum of DNA binding sites of a specific transcription factor is referred to as its cistrome. DNA binding sites also encompasses the targets of other proteins, like restriction enzymes, site-specific recombinases and methyltransferases.
Chromatin Interaction Analysis by Paired-End Tag Sequencing is a technique that incorporates chromatin immunoprecipitation (ChIP)-based enrichment, chromatin proximity ligation, Paired-End Tags, and High-throughput sequencing to determine de novo long-range chromatin interactions genome-wide.
Genes can be regulated by regions far from the promoter such as regulatory elements, insulators and boundary elements, and transcription-factor binding sites (TFBS). Uncovering the interplay between regulatory regions and gene coding regions is essential for understanding the mechanisms governing gene regulation in health and disease. ChIA-PET can be used to identify unique, functional chromatin interactions between distal and proximal regulatory transcription-factor binding sites and the promoters of the genes they interact with.
Peak calling is a computational method used to identify areas in a genome that have been enriched with aligned reads as a consequence of performing a ChIP-sequencing or MeDIP-seq experiment. These areas are those where a protein interacts with DNA. When the protein is a transcription factor, the enriched area is its transcription factor binding site (TFBS). Popular software programs include MACS. Wilbanks and colleagues is a survey of the ChIP-seq peak callers, and Bailey et al. is a description of practical guidelines for peak calling in ChIP-seq data.

ChIP-exo is a chromatin immunoprecipitation based method for mapping the locations at which a protein of interest binds to the genome. It is a modification of the ChIP-seq protocol, improving the resolution of binding sites from hundreds of base pairs to almost one base pair. It employs the use of exonucleases to degrade strands of the protein-bound DNA in the 5'-3' direction to within a small number of nucleotides of the protein binding site. The nucleotides of the exonuclease-treated ends are determined using some combination of DNA sequencing, microarrays, and PCR. These sequences are then mapped to the genome to identify the locations on the genome at which the protein binds.
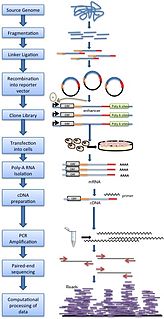
STARR-seq is a novel method to assay enhancer activity for millions of candidates from arbitrary sources of DNA. It is used to identify the sequences that act as transcriptional enhancers in a direct, quantitative, and genome-wide manner.
Chem-seq is a technique that is used to map genome-wide interactions between small molecules and their protein targets in the chromatin of eukaryotic cell nuclei. The method employs chemical affinity capture coupled with massively parallel DNA sequencing to identify genomic sites where small molecules interact with their target proteins or DNA. It was first described by Lars Anders et al. in the January, 2014 issue of "Nature Biotechnology".
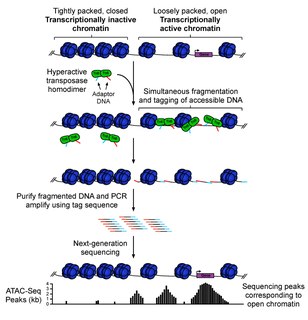
ATAC-seq is a technique used in molecular biology to assess genome-wide chromatin accessibility(1). In 2013, the technique was first described as an alternative advanced method for MNase-seq, FAIRE-seq and DNAse-seq (1). ATAC-seq is an emerging technique that’s gaining popularity among researchers from diverse backgrounds as it aids in a fast and sensitive analysis of the epigenome compared to DNase-seq or MNase-seq (2,3,4). The applications of ATAC-seq in enhancing the functional genomics field have been explored in recent literature in hopes to understand epigenetic regulation in the context of disease development and cell differentiation. Indeed, ATAC-seq is becoming an essential tool in epigenetics and genome-regulation research and a standard part of epigenetic analysis. It has been successfully adapted to efficiently identify open chromatin and identify regulatory elements across the genome.

Single cell epigenomics is the study of epigenomics in individual cells by single cell sequencing. Since 2013, methods have been created including whole-genome single-cell bisulfite sequencing to measure DNA methylation, whole-genome ChIP-sequencing to measure histone modifications, whole-genome ATAC-seq to measure chromatin accessibility and chromosome conformation capture.
CUT&RUN-sequencing, also known as cleavage under targets and release using nuclease, is a method used to analyze protein interactions with DNA. CUT&RUN-sequencing combines antibody-targeted controlled cleavage by micrococcal nuclease with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global DNA binding sites precisely for any protein of interest. Currently, ChIP-Seq is the most common technique utilized to study protein–DNA relations, however, it suffers from a number of practical and economical limitations that CUT&RUN-Sequencing does not.


