
Ichthyosis vulgaris is a skin disorder causing dry, scaly skin. It is the most common form of ichthyosis, affecting around 1 in 250 people. For this reason it is known as common ichthyosis. It is usually an autosomal dominant inherited disease, although a rare non-heritable version called acquired ichthyosis exists.

Primary familial brain calcification (PFBC), also known as familial idiopathic basal ganglia calcification (FIBGC) and Fahr's disease, is a rare, genetically dominant, inherited neurological disorder characterized by abnormal deposits of calcium in areas of the brain that control movement. Through the use of CT scans, calcifications are seen primarily in the basal ganglia and in other areas such as the cerebral cortex.

Z-DNA is one of the many possible double helical structures of DNA. It is a left-handed double helical structure in which the helix winds to the left in a zigzag pattern, instead of to the right, like the more common B-DNA form. Z-DNA is thought to be one of three biologically active double-helical structures along with A-DNA and B-DNA.

Incontinentia pigmenti (IP) is a rare X-linked dominant genetic disorder that affects the skin, hair, teeth, nails and central nervous system. It is named from its appearance under a microscope.

Palmoplantar keratodermas are a heterogeneous group of disorders characterized by abnormal thickening of the stratum corneum of the palms and soles.
Dyschromatosis universalis hereditaria is a type of pigmentation disorder of the skin. It is characterized by dark and light spots formed like lace in a generalized distribution.

Epidermolysis bullosa simplex (EBS) is a disorder resulting from mutations in the genes encoding keratin 5 or keratin 14.

Naegeli–Franceschetti–Jadassohn syndrome (NFJS), also known as chromatophore nevus of Naegeli and Naegeli syndrome, is a rare autosomal dominant form of ectodermal dysplasia, characterized by reticular skin pigmentation, diminished function of the sweat glands, the absence of teeth and hyperkeratosis of the palms and soles. One of the most striking features is the absence of fingerprint lines on the fingers.
Dermatopathia pigmentosa reticularis(DPR) is a rare, autosomal dominant congenital disorder that is a form of ectodermal dysplasia. Dermatopathia pigmentosa reticularis is composed of the triad of generalized reticulate hyperpigmentation, noncicatricial alopecia, and onychodystrophy. DPR is a non life-threatening disease that largely affects the skin, hair, and nails. It has also been identified as a keratin disorder. Historically, as of 1992, only 10 cases had been described in world literature; however, due to recent advances in genetic analysis, five additional families studied in 2006 have been added to the short list of confirmed cases.

The double-stranded RNA-specific adenosine deaminase enzyme family are encoded by the ADAR family genes. ADAR stands for adenosine deaminase acting on RNA. This article focuses on the ADAR proteins; This article details the evolutionary history, structure, function, mechanisms and importance of all proteins within this family.

ATP-binding cassette super-family B member 6, mitochondrial is a protein that in humans is encoded by the ABCB6 gene.

Gerodermia osteodysplastica (GO) is a rare autosomal recessive connective tissue disorder included in the spectrum of cutis laxa syndromes.
Reticulate acropigmentation of Kitamura is a type of pigmentation disorder of the skin. It presents with coloured freckle-like and slightly depressed flat spots arranged in a lace-like pattern on the backs of hands and feet. It tends to occur in skin folds of teenagers and in early adulthood, and darkens over time.
Peeling skin syndrome is an autosomal recessive disorder characterized by lifelong peeling of the stratum corneum, and may be associated with pruritus, short stature, and easily removed anagen hair.
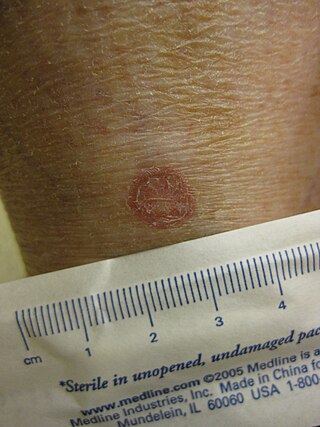
Porokeratosis is a specific disorder of keratinization that is characterized histologically by the presence of a cornoid lamella, a thin column of closely stacked, parakeratotic cells extending through the stratum corneum with a thin or absent granular layer.
Cryopyrin-associated periodic syndrome (CAPS) is a group of rare, heterogeneous autoinflammatory disease characterized by interleukin 1β-mediated systemic inflammation and clinical symptoms involving skin, joints, central nervous system, and eyes. It encompasses a spectrum of three clinically overlapping autoinflammatory syndromes including familial cold autoinflammatory syndrome, the Muckle–Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disease that were originally thought to be distinct entities, but in fact share a single genetic mutation and pathogenic pathway, and keratoendotheliitis fugax hereditaria in which the autoinflammatory symptoms affect only the anterior segment of the eye.

Aicardi–Goutières syndrome (AGS), which is completely distinct from the similarly named Aicardi syndrome, is a rare, usually early onset childhood, inflammatory disorder most typically affecting the brain and the skin. The majority of affected individuals experience significant intellectual and physical problems, although this is not always the case. The clinical features of AGS can mimic those of in utero acquired infection, and some characteristics of the condition also overlap with the autoimmune disease systemic lupus erythematosus (SLE). Following an original description of eight cases in 1984, the condition was first referred to as 'Aicardi–Goutières syndrome' (AGS) in 1992, and the first international meeting on AGS was held in Pavia, Italy, in 2001.
Acro–dermato–ungual–lacrimal–tooth syndrome is a rare genetic disease. It is an autosomal dominant form of ectodermal dysplasia, a group of disorders that affects the hair, teeth, nails, sweat glands, and extremities. The syndrome arises from a mutation in the TP63 gene. This disease was previously thought to be a form of ectrodactyly–ectodermal dysplasia–cleft syndrome (EEC), but was classified as a different disease in 1993 by Propping and Zerres.
Keratoendotheliitis fugax hereditaria is an autosomal dominantly inherited disease of the cornea, caused by a point mutation in cryopyrin that in humans is encoded by the NLRP3 gene located on the long arm of chromosome 1.
