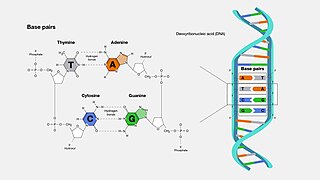
A base pair (bp) is a fundamental unit of double-stranded nucleic acids consisting of two nucleobases bound to each other by hydrogen bonds. They form the building blocks of the DNA double helix and contribute to the folded structure of both DNA and RNA. Dictated by specific hydrogen bonding patterns, "Watson–Crick" base pairs allow the DNA helix to maintain a regular helical structure that is subtly dependent on its nucleotide sequence. The complementary nature of this based-paired structure provides a redundant copy of the genetic information encoded within each strand of DNA. The regular structure and data redundancy provided by the DNA double helix make DNA well suited to the storage of genetic information, while base-pairing between DNA and incoming nucleotides provides the mechanism through which DNA polymerase replicates DNA and RNA polymerase transcribes DNA into RNA. Many DNA-binding proteins can recognize specific base-pairing patterns that identify particular regulatory regions of genes.

In molecular biology, a stop codon is a codon that signals the termination of the translation process of the current protein. Most codons in messenger RNA correspond to the addition of an amino acid to a growing polypeptide chain, which may ultimately become a protein; stop codons signal the termination of this process by binding release factors, which cause the ribosomal subunits to disassociate, releasing the amino acid chain.
Molecular evolution is the process of change in the sequence composition of cellular molecules such as DNA, RNA, and proteins across generations. The field of molecular evolution uses principles of evolutionary biology and population genetics to explain patterns in these changes. Major topics in molecular evolution concern the rates and impacts of single nucleotide changes, neutral evolution vs. natural selection, origins of new genes, the genetic nature of complex traits, the genetic basis of speciation, the evolution of development, and ways that evolutionary forces influence genomic and phenotypic changes.
The coding region of a gene, also known as the coding sequence(CDS), is the portion of a gene's DNA or RNA that codes for a protein. Studying the length, composition, regulation, splicing, structures, and functions of coding regions compared to non-coding regions over different species and time periods can provide a significant amount of important information regarding gene organization and evolution of prokaryotes and eukaryotes. This can further assist in mapping the human genome and developing gene therapy.

The CpG sites or CG sites are regions of DNA where a cytosine nucleotide is followed by a guanine nucleotide in the linear sequence of bases along its 5' → 3' direction. CpG sites occur with high frequency in genomic regions called CpG islands.

Codon usage bias refers to differences in the frequency of occurrence of synonymous codons in coding DNA. A codon is a series of three nucleotides that encodes a specific amino acid residue in a polypeptide chain or for the termination of translation.
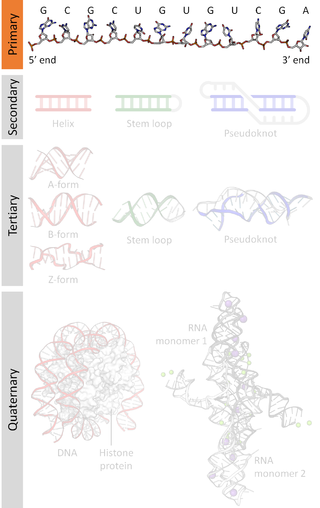
A nucleic acid sequence is a succession of bases within the nucleotides forming alleles within a DNA or RNA (GACU) molecule. This succession is denoted by a series of a set of five different letters that indicate the order of the nucleotides. By convention, sequences are usually presented from the 5' end to the 3' end. For DNA, with its double helix, there are two possible directions for the notated sequence; of these two, the sense strand is used. Because nucleic acids are normally linear (unbranched) polymers, specifying the sequence is equivalent to defining the covalent structure of the entire molecule. For this reason, the nucleic acid sequence is also termed the primary structure.

Molecular genetics is a branch of biology that addresses how differences in the structures or expression of DNA molecules manifests as variation among organisms. Molecular genetics often applies an "investigative approach" to determine the structure and/or function of genes in an organism's genome using genetic screens.

Chargaff's rules state that in the DNA of any species and any organism, the amount of guanine should be equal to the amount of cytosine and the amount of adenine should be equal to the amount of thymine. Further, a 1:1 stoichiometric ratio of purine and pyrimidine bases should exist. This pattern is found in both strands of the DNA. They were discovered by Austrian-born chemist Erwin Chargaff in the late 1940s.
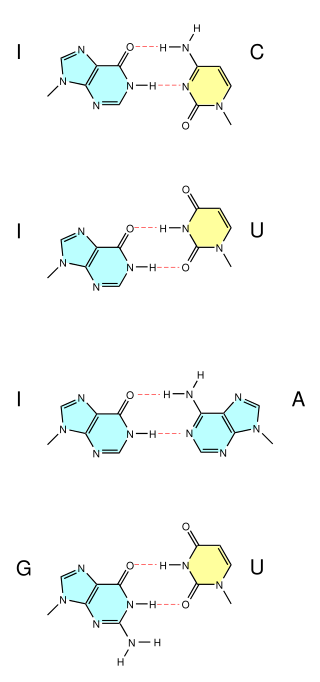
A wobble base pair is a pairing between two nucleotides in RNA molecules that does not follow Watson-Crick base pair rules. The four main wobble base pairs are guanine-uracil (G-U), hypoxanthine-uracil (I-U), hypoxanthine-adenine (I-A), and hypoxanthine-cytosine (I-C). In order to maintain consistency of nucleic acid nomenclature, "I" is used for hypoxanthine because hypoxanthine is the nucleobase of inosine; nomenclature otherwise follows the names of nucleobases and their corresponding nucleosides. The thermodynamic stability of a wobble base pair is comparable to that of a Watson-Crick base pair. Wobble base pairs are fundamental in RNA secondary structure and are critical for the proper translation of the genetic code.
Gene conversion is the process by which one DNA sequence replaces a homologous sequence such that the sequences become identical after the conversion event. Gene conversion can be either allelic, meaning that one allele of the same gene replaces another allele, or ectopic, meaning that one paralogous DNA sequence converts another.
Nucleic acid thermodynamics is the study of how temperature affects the nucleic acid structure of double-stranded DNA (dsDNA). The melting temperature (Tm) is defined as the temperature at which half of the DNA strands are in the random coil or single-stranded (ssDNA) state. Tm depends on the length of the DNA molecule and its specific nucleotide sequence. DNA, when in a state where its two strands are dissociated, is referred to as having been denatured by the high temperature.

Bisulfitesequencing (also known as bisulphite sequencing) is the use of bisulfite treatment of DNA before routine sequencing to determine the pattern of methylation. DNA methylation was the first discovered epigenetic mark, and remains the most studied. In animals it predominantly involves the addition of a methyl group to the carbon-5 position of cytosine residues of the dinucleotide CpG, and is implicated in repression of transcriptional activity.

In bioinformatics, k-mers are substrings of length contained within a biological sequence. Primarily used within the context of computational genomics and sequence analysis, in which k-mers are composed of nucleotides, k-mers are capitalized upon to assemble DNA sequences, improve heterologous gene expression, identify species in metagenomic samples, and create attenuated vaccines. Usually, the term k-mer refers to all of a sequence's subsequences of length , such that the sequence AGAT would have four monomers, three 2-mers, two 3-mers and one 4-mer (AGAT). More generally, a sequence of length will have k-mers and total possible k-mers, where is number of possible monomers.
In genetics, an isochore is a large region of genomic DNA with a high degree of uniformity in GC content; that is, guanine (G) and cytosine (C) bases. The distribution of bases within a genome is non-random: different regions of the genome have different amounts of G-C base pairs, such that regions can be classified and identified by the proportion of G-C base pairs they contain.

Nucleic acid secondary structure is the basepairing interactions within a single nucleic acid polymer or between two polymers. It can be represented as a list of bases which are paired in a nucleic acid molecule. The secondary structures of biological DNAs and RNAs tend to be different: biological DNA mostly exists as fully base paired double helices, while biological RNA is single stranded and often forms complex and intricate base-pairing interactions due to its increased ability to form hydrogen bonds stemming from the extra hydroxyl group in the ribose sugar.

Genome evolution is the process by which a genome changes in structure (sequence) or size over time. The study of genome evolution involves multiple fields such as structural analysis of the genome, the study of genomic parasites, gene and ancient genome duplications, polyploidy, and comparative genomics. Genome evolution is a constantly changing and evolving field due to the steadily growing number of sequenced genomes, both prokaryotic and eukaryotic, available to the scientific community and the public at large.

A compositional domain in genetics is a region of DNA with a distinct guanine (G) and cytosine (C) G-C and C-G content. The homogeneity of compositional domains is compared to that of the chromosome on which they reside. As such, compositional domains can be homogeneous or nonhomogeneous domains. Compositionally homogeneous domains that are sufficiently long are termed isochores or isochoric domains.

GC skew is when the nucleotides guanine and cytosine are over- or under-abundant in a particular region of DNA or RNA. GC skew is also a statistical method for measuring strand-specific guanine overrepresentation.
The invertebrate mitochondrial code is a genetic code used by the mitochondrial genome of invertebrates. Mitochondria contain their own DNA and reproduce independently from their host cell. Variation in translation of the mitochondrial genetic code occurs when DNA codons result in non-standard amino acids has been identified in invertebrates, most notably arthropods. This variation has been helpful as a tool to improve upon the phylogenetic tree of invertebrates, like flatworms.



