An allele, or allelomorph, is a variant of the sequence of nucleotides at a particular location, or locus, on a DNA molecule.
Genetic drift, also known as random genetic drift, allelic drift or the Wright effect, is the change in the frequency of an existing gene variant (allele) in a population due to random chance.

The neutral theory of molecular evolution holds that most evolutionary changes occur at the molecular level, and most of the variation within and between species are due to random genetic drift of mutant alleles that are selectively neutral. The theory applies only for evolution at the molecular level, and is compatible with phenotypic evolution being shaped by natural selection as postulated by Charles Darwin.
Fitness is a quantitative representation of individual reproductive success. It is also equal to the average contribution to the gene pool of the next generation, made by the same individuals of the specified genotype or phenotype. Fitness can be defined either with respect to a genotype or to a phenotype in a given environment or time. The fitness of a genotype is manifested through its phenotype, which is also affected by the developmental environment. The fitness of a given phenotype can also be different in different selective environments.
Population genetics is a subfield of genetics that deals with genetic differences within and among populations, and is a part of evolutionary biology. Studies in this branch of biology examine such phenomena as adaptation, speciation, and population structure.
Allele frequency, or gene frequency, is the relative frequency of an allele at a particular locus in a population, expressed as a fraction or percentage. Specifically, it is the fraction of all chromosomes in the population that carry that allele over the total population or sample size. Microevolution is the change in allele frequencies that occurs over time within a population.
Genetic linkage is the tendency of DNA sequences that are close together on a chromosome to be inherited together during the meiosis phase of sexual reproduction. Two genetic markers that are physically near to each other are unlikely to be separated onto different chromatids during chromosomal crossover, and are therefore said to be more linked than markers that are far apart. In other words, the nearer two genes are on a chromosome, the lower the chance of recombination between them, and the more likely they are to be inherited together. Markers on different chromosomes are perfectly unlinked, although the penetrance of potentially deleterious alleles may be influenced by the presence of other alleles, and these other alleles may be located on other chromosomes than that on which a particular potentially deleterious allele is located.

Genetic diversity is the total number of genetic characteristics in the genetic makeup of a species, it ranges widely from the number of species to differences within species and can be attributed to the span of survival for a species. It is distinguished from genetic variability, which describes the tendency of genetic characteristics to vary.
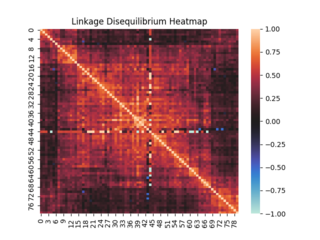
In population genetics, linkage disequilibrium (LD) is a measure of non-random association between segments of DNA (alleles) at different positions on the chromosome (loci) in a given population based on a comparison between the frequency at which two alleles are detected together at the same loci and the frequencies at which each allele is detected at that loci overall, whether it occurs with or without the other allele of interest. Loci are said to be in linkage disequilibrium when the frequency of being detected together is higher or lower than expected if the loci were independent and associated randomly.
Nucleotide diversity is a concept in molecular genetics which is used to measure the degree of polymorphism within a population.
The effective population size (Ne) is size of an idealised population would experience the same rate of genetic drift or increase in inbreeding as in the real population. Idealised populations are based on unrealistic but convenient assumptions including random mating, simultaneous birth of each new generation, constant population size. For most quantities of interest and most real populations, Ne is smaller than the census population size N of a real population. The same population may have multiple effective population sizes for different properties of interest, including genetic drift and inbreeding.

Conservation genetics is an interdisciplinary subfield of population genetics that aims to understand the dynamics of genes in a population for the purpose of natural resource management, conservation of genetic diversity, and the prevention of species extinction. Scientists involved in conservation genetics come from a variety of fields including population genetics, research in natural resource management, molecular ecology, molecular biology, evolutionary biology, and systematics. The genetic diversity within species is one of the three fundamental components of biodiversity, so it is an important consideration in the wider field of conservation biology.
Genetic load is the difference between the fitness of an average genotype in a population and the fitness of some reference genotype, which may be either the best present in a population, or may be the theoretically optimal genotype. The average individual taken from a population with a low genetic load will generally, when grown in the same conditions, have more surviving offspring than the average individual from a population with a high genetic load. Genetic load can also be seen as reduced fitness at the population level compared to what the population would have if all individuals had the reference high-fitness genotype. High genetic load may put a population in danger of extinction.
Genetic hitchhiking, also called genetic draft or the hitchhiking effect, is when an allele changes frequency not because it itself is under natural selection, but because it is near another gene that is undergoing a selective sweep and that is on the same DNA chain. When one gene goes through a selective sweep, any other nearby polymorphisms that are in linkage disequilibrium will tend to change their allele frequencies too. Selective sweeps happen when newly appeared mutations are advantageous and increase in frequency. Neutral or even slightly deleterious alleles that happen to be close by on the chromosome 'hitchhike' along with the sweep. In contrast, effects on a neutral locus due to linkage disequilibrium with newly appeared deleterious mutations are called background selection. Both genetic hitchhiking and background selection are stochastic (random) evolutionary forces, like genetic drift.
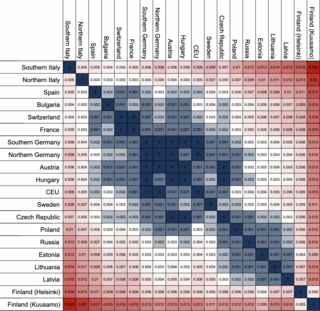
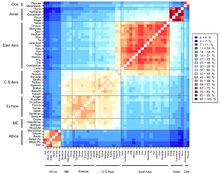
The fixation index (FST) is a measure of population differentiation due to genetic structure. It is frequently estimated from genetic polymorphism data, such as single-nucleotide polymorphisms (SNP) or microsatellites. Developed as a special case of Wright's F-statistics, it is one of the most commonly used statistics in population genetics. Its values range from 0 to 1, with 0.15 being substantially differentiated and 1 being complete differentiation.

Masatoshi Nei was a Japanese-born American evolutionary biologist.
In population genetics, fixation is the change in a gene pool from a situation where there exists at least two variants of a particular gene (allele) in a given population to a situation where only one of the alleles remains. That is, the allele becomes fixed. In the absence of mutation or heterozygote advantage, any allele must eventually either be lost completely from the population, or fixed, i.e. permanently established at 100% frequency in the population. Whether a gene will ultimately be lost or fixed is dependent on selection coefficients and chance fluctuations in allelic proportions. Fixation can refer to a gene in general or particular nucleotide position in the DNA chain (locus).
Population structure is the presence of a systematic difference in allele frequencies between subpopulations. In a randomly mating population, allele frequencies are expected to be roughly similar between groups. However, mating tends to be non-random to some degree, causing structure to arise. For example, a barrier like a river can separate two groups of the same species and make it difficult for potential mates to cross; if a mutation occurs, over many generations it can spread and become common in one subpopulation while being completely absent in the other.
The Infinite sites model (ISM) is a mathematical model of molecular evolution first proposed by Motoo Kimura in 1969. Like other mutation models, the ISM provides a basis for understanding how mutation develops new alleles in DNA sequences. Using allele frequencies, it allows for the calculation of heterozygosity, or genetic diversity, in a finite population and for the estimation of genetic distances between populations of interest.
This glossary of genetics and evolutionary biology is a list of definitions of terms and concepts used in the study of genetics and evolutionary biology, as well as sub-disciplines and related fields, with an emphasis on classical genetics, quantitative genetics, population biology, phylogenetics, speciation, and systematics. Overlapping and related terms can be found in Glossary of cellular and molecular biology, Glossary of ecology, and Glossary of biology.