
Porphyria is a group of disorders in which substances called porphyrins build up in the body, adversely affecting the skin or nervous system. The types that affect the nervous system are also known as acute porphyria, as symptoms are rapid in onset and short in duration. Symptoms of an attack include abdominal pain, chest pain, vomiting, confusion, constipation, fever, high blood pressure, and high heart rate. The attacks usually last for days to weeks. Complications may include paralysis, low blood sodium levels, and seizures. Attacks may be triggered by alcohol, smoking, hormonal changes, fasting, stress, or certain medications. If the skin is affected, blisters or itching may occur with sunlight exposure.

Heme, or haem, is a ring-shaped iron-containing molecular component of hemoglobin, which is necessary to bind oxygen in the bloodstream. It is composed of four pyrrole rings with 2 vinyl and 2 propionic acid side chains. Heme is biosynthesized in both the bone marrow and the liver.

Hereditary coproporphyria (HCP) is a disorder of heme biosynthesis, classified as an acute hepatic porphyria. HCP is caused by a deficiency of the enzyme coproporphyrinogen oxidase, coded for by the CPOX gene, and is inherited in an autosomal dominant fashion, although homozygous individuals have been identified. Unlike acute intermittent porphyria, individuals with HCP can present with cutaneous findings similar to those found in porphyria cutanea tarda in addition to the acute attacks of abdominal pain, vomiting and neurological dysfunction characteristic of acute porphyrias. Like other porphyrias, attacks of HCP can be induced by certain drugs, environmental stressors or diet changes. Biochemical and molecular testing can be used to narrow down the diagnosis of a porphyria and identify the specific genetic defect. Overall, porphyrias are rare diseases. The combined incidence for all forms of the disease has been estimated at 1:20,000. The exact incidence of HCP is difficult to determine, due to its reduced penetrance.

Variegate porphyria, also known by several other names, is an autosomal dominant porphyria that can have acute symptoms along with symptoms that affect the skin. The disorder results from low levels of the enzyme responsible for the seventh step in heme production. Heme is a vital molecule for all of the body's organs. It is a component of hemoglobin, the molecule that carries oxygen in the blood.

Porphyria cutanea tarda is the most common subtype of porphyria. The disease is named because it is a porphyria that often presents with skin manifestations later in life. The disorder results from low levels of the enzyme responsible for the fifth step in heme production. Heme is a vital molecule for all of the body's organs. It is a component of hemoglobin, the molecule that carries oxygen in the blood.

Erythropoietic protoporphyria is a form of porphyria, which varies in severity and can be very painful. It arises from a deficiency in the enzyme ferrochelatase, leading to abnormally high levels of protoporphyrin in the red blood cells (erythrocytes), plasma, skin, and liver. The severity varies significantly from individual to individual.
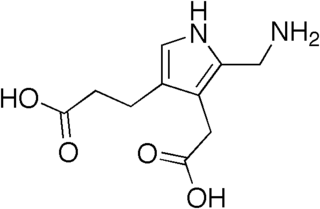
Aminolevulinic acid synthase (ALA synthase, ALAS, or delta-aminolevulinic acid synthase) is an enzyme (EC 2.3.1.37) that catalyzes the synthesis of δ-aminolevulinic acid (ALA) the first common precursor in the biosynthesis of all tetrapyrroles such as hemes, cobalamins and chlorophylls. The reaction is as follows:
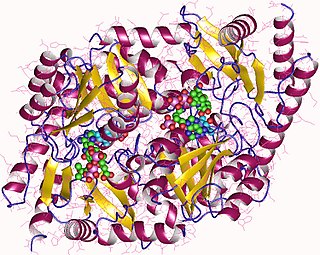
Acute intermittent porphyria (AIP) is a rare metabolic disorder affecting the production of heme resulting from a deficiency of the enzyme porphobilinogen deaminase. It is the most common of the acute porphyrias.

Protoporphyrinogen oxidase or protox is an enzyme that in humans is encoded by the PPOX gene.

Uroporphyrinogen III decarboxylase is an enzyme that in humans is encoded by the UROD gene.

GATA-binding factor 1 or GATA-1 is the founding member of the GATA family of transcription factors. This protein is widely expressed throughout vertebrate species. In humans and mice, it is encoded by the GATA1 and Gata1 genes, respectively. These genes are located on the X chromosome in both species.

Uroporphyrinogen III synthase is an enzyme involved in the metabolism of the cyclic tetrapyrrole compound porphyrin. It is involved in the conversion of hydroxymethyl bilane into uroporphyrinogen III. This enzyme catalyses the inversion of the final pyrrole unit of the linear tetrapyrrole molecule, linking it to the first pyrrole unit, thereby generating a large macrocyclic structure, uroporphyrinogen III. The enzyme folds into two alpha/beta domains connected by a beta-ladder, the active site being located between the two domains.

Uroporphyrinogen III is a tetrapyrrole, the first macrocyclic intermediate in the biosynthesis of heme, chlorophyll, vitamin B12, and siroheme. It is a colorless compound, like other porphyrinogens.
Erythropoietic porphyria is a type of porphyria associated with erythropoietic cells. In erythropoietic porphyrias, the enzyme deficiency occurs in the red blood cells.
Hepatoerythropoietic porphyria is a very rare form of hepatic porphyria caused by a disorder in both genes which code Uroporphyrinogen III decarboxylase (UROD).

Uroporphyrinogen I is an isomer of uroporphyrinogen III, a metabolic intermediate in the biosynthesis of heme. A type of porphyria is caused by production of uroporphyrinogen I instead of III.

Coproporphyrinogen I is an isomer of coproporphyrinogen III, a metabolic intermediate in the normal biosynthesis of heme. The compound is not normally produced by the human body; its production and accumulation causes a type of porphyria.
Congenital hemolytic anemia (CHA) is a diverse group of rare hereditary conditions marked by decreased life expectancy and premature removal of erythrocytes from blood flow. Defects in erythrocyte membrane proteins and red cell enzyme metabolism, as well as changes at the level of erythrocyte precursors, lead to impaired bone marrow erythropoiesis. CAH is distinguished by variable anemia, chronic extravascular hemolysis, decreased erythrocyte life span, splenomegaly, jaundice, biliary lithiasis, and iron overload. Immune-mediated mechanisms may play a role in the pathogenesis of these uncommon diseases, despite the paucity of data regarding the immune system's involvement in CHAs.

Aminolevulinic acid dehydratase deficiency porphyria is an extremely rare autosomal recessive metabolic disorder that results from inappropriately low levels of the enzyme delta-aminolevulinic acid dehydratase (ALAD), which is required for normal heme synthesis. This deficiency results in the accumulation of a toxic metabolic precursor in the heme synthesis pathway called aminolevulinic acid (ALA). Lead poisoning can also disrupt ALAD and result in elevated ALA causing the same symptoms. Heme is a component of hemoglobin which carries oxygen in red blood cells.
Harderoporphyria is a rare disorder of heme biosynthesis, inherited in an autosomal recessive manner caused by specific mutations in the CPOX gene. Mutations in CPOX usually cause hereditary coproporphyria, an acute hepatic porphyria, however the K404E mutation in a homozygous or compound heterozygous state with a null allele cause the more severe harderoporphyria. Harderoporphyria is the first known metabolic disorder where the disease phenotype depended on the type and location of the mutations in a gene associated with multiple disorders.


