An epitope, also known as antigenic determinant, is the part of an antigen that is recognized by the immune system, specifically by antibodies, B cells, or T cells. The part of an antibody that binds to the epitope is called a paratope. Although epitopes are usually non-self proteins, sequences derived from the host that can be recognized are also epitopes.

Flow cytometry (FC) is a technique used to detect and measure physical and chemical characteristics of a population of cells or particles.

Immunofluorescence(IF) is a light microscopy-based technique that allows detection and localization of a wide variety of target biomolecules within a cell or tissue at a quantitative level. The technique utilizes the binding specificity of antibodies and antigens. The specific region an antibody recognizes on an antigen is called an epitope. Several antibodies can recognize the same epitope but differ in their binding affinity. The antibody with the higher affinity for a specific epitope will surpass antibodies with a lower affinity for the same epitope.
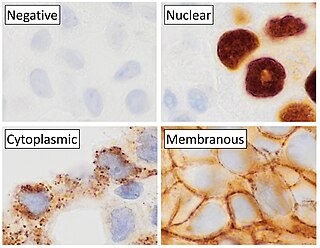
Immunohistochemistry (IHC) is a form of immunostaining. It involves the process of selectively identifying antigens (proteins) in cells and tissue, by exploiting the principle of antibodies binding specifically to antigens in biological tissues. Albert Hewett Coons, Ernest Berliner, Norman Jones and Hugh J Creech was the first to develop immunofluorescence in 1941. This led to the later development of immunohistochemistry.

A fluorescence microscope is an optical microscope that uses fluorescence instead of, or in addition to, scattering, reflection, and attenuation or absorption, to study the properties of organic or inorganic substances. "Fluorescence microscope" refers to any microscope that uses fluorescence to generate an image, whether it is a simple set up like an epifluorescence microscope or a more complicated design such as a confocal microscope, which uses optical sectioning to get better resolution of the fluorescence image.

Hoechst stains are part of a family of blue fluorescent dyes used to stain DNA. These bis-benzimides were originally developed by Hoechst AG, which numbered all their compounds so that the dye Hoechst 33342 is the 33,342nd compound made by the company. There are three related Hoechst stains: Hoechst 33258, Hoechst 33342, and Hoechst 34580. The dyes Hoechst 33258 and Hoechst 33342 are the ones most commonly used and they have similar excitation–emission spectra.
Cyanines, also referred to as tetramethylindo(di)-carbocyanines are a synthetic dye family belonging to the polymethine group. Although the name derives etymologically from terms for shades of blue, the cyanine family covers the electromagnetic spectrum from near IR to UV.
Bacterial display is a protein engineering technique used for in vitro protein evolution. Libraries of polypeptides displayed on the surface of bacteria can be screened using flow cytometry or iterative selection procedures (biopanning). This protein engineering technique allows us to link the function of a protein with the gene that encodes it. Bacterial display can be used to find target proteins with desired properties and can be used to make affinity ligands which are cell-specific. This system can be used in many applications including the creation of novel vaccines, the identification of enzyme substrates and finding the affinity of a ligand for its target protein.

Stimulated emission depletion (STED) microscopy is one of the techniques that make up super-resolution microscopy. It creates super-resolution images by the selective deactivation of fluorophores, minimizing the area of illumination at the focal point, and thus enhancing the achievable resolution for a given system. It was developed by Stefan W. Hell and Jan Wichmann in 1994, and was first experimentally demonstrated by Hell and Thomas Klar in 1999. Hell was awarded the Nobel Prize in Chemistry in 2014 for its development. In 1986, V.A. Okhonin had patented the STED idea. This patent was unknown to Hell and Wichmann in 1994.
High-content screening (HCS), also known as high-content analysis (HCA) or cellomics, is a method that is used in biological research and drug discovery to identify substances such as small molecules, peptides, or RNAi that alter the phenotype of a cell in a desired manner. Hence high content screening is a type of phenotypic screen conducted in cells involving the analysis of whole cells or components of cells with simultaneous readout of several parameters. HCS is related to high-throughput screening (HTS), in which thousands of compounds are tested in parallel for their activity in one or more biological assays, but involves assays of more complex cellular phenotypes as outputs. Phenotypic changes may include increases or decreases in the production of cellular products such as proteins and/or changes in the morphology of the cell. Hence HCA typically involves automated microscopy and image analysis. Unlike high-content analysis, high-content screening implies a level of throughput which is why the term "screening" differentiates HCS from HCA, which may be high in content but low in throughput.

Cytomics is the study of cell biology (cytology) and biochemistry in cellular systems at the single cell level. It combines all the bioinformatic knowledge to attempt to understand the molecular architecture and functionality of the cell system (Cytome). Much of this is achieved by using molecular and microscopic techniques that allow the various components of a cell to be visualised as they interact in vivo.

7-Aminoactinomycin D (7-AAD) is a fluorescent chemical compound with a strong affinity for DNA. It is used as a fluorescent marker for DNA in fluorescence microscopy and flow cytometry. It intercalates in double-stranded DNA, with a high affinity for GC-rich regions, making it useful for chromosome banding studies.

Cytometry is the measurement of number and characteristics of cells. Variables that can be measured by cytometric methods include cell size, cell count, cell morphology, cell cycle phase, DNA content, and the existence or absence of specific proteins on the cell surface or in the cytoplasm. Cytometry is used to characterize and count blood cells in common blood tests such as the complete blood count. In a similar fashion, cytometry is also used in cell biology research and in medical diagnostics to characterize cells in a wide range of applications associated with diseases such as cancer and AIDS.

Robert F. Murphy is Ray and Stephanie Lane Professor of Computational Biology Emeritus and Director of the M.S. Program in Automated Science at Carnegie Mellon University. Prior to his retirement in May 2021, he was the Ray and Stephanie Lane Professor of Computational Biology as well as Professor of Biological Sciences, Biomedical Engineering, and Machine Learning. He was founding Director of the Center for Bioimage Informatics at Carnegie Mellon and founded the Joint CMU-Pitt Ph.D. Program in Computational Biology. He also founded the Computational Biology Department at Carnegie Mellon University and served as its head from 2009 to 2020.
The Human Protein Atlas (HPA) is a Swedish-based program started in 2003 with the aim to map all the human proteins in cells, tissues and organs using integration of various omics technologies, including antibody-based imaging, mass spectrometry-based proteomics, transcriptomics and systems biology. All the data in the knowledge resource is open access to allow scientists both in academia and industry to freely access the data for exploration of the human proteome. In June 2023, version 23 was launched where a new Interaction section was introduced containing human protein-protein interaction networks for more than 11,000 genes that will add new aspects in terms of protein function.

In the field of cellular biology, single-cell analysis and subcellular analysis is the study of genomics, transcriptomics, proteomics, metabolomics and cell–cell interactions at the single cell level. The concept of single-cell analysis originated in the 1970s. Before the discovery of heterogeneity, single-cell analysis mainly referred to the analysis or manipulation of an individual cell in a bulk population of cells at a particular condition using optical or electronic microscope. To date, due to the heterogeneity seen in both eukaryotic and prokaryotic cell populations, analyzing a single cell makes it possible to discover mechanisms not seen when studying a bulk population of cells. Technologies such as fluorescence-activated cell sorting (FACS) allow the precise isolation of selected single cells from complex samples, while high throughput single cell partitioning technologies, enable the simultaneous molecular analysis of hundreds or thousands of single unsorted cells; this is particularly useful for the analysis of transcriptome variation in genotypically identical cells, allowing the definition of otherwise undetectable cell subtypes. The development of new technologies is increasing our ability to analyze the genome and transcriptome of single cells, as well as to quantify their proteome and metabolome. Mass spectrometry techniques have become important analytical tools for proteomic and metabolomic analysis of single cells. Recent advances have enabled quantifying thousands of protein across hundreds of single cells, and thus make possible new types of analysis. In situ sequencing and fluorescence in situ hybridization (FISH) do not require that cells be isolated and are increasingly being used for analysis of tissues.
Flow cytometry bioinformatics is the application of bioinformatics to flow cytometry data, which involves storing, retrieving, organizing and analyzing flow cytometry data using extensive computational resources and tools. Flow cytometry bioinformatics requires extensive use of and contributes to the development of techniques from computational statistics and machine learning. Flow cytometry and related methods allow the quantification of multiple independent biomarkers on large numbers of single cells. The rapid growth in the multidimensionality and throughput of flow cytometry data, particularly in the 2000s, has led to the creation of a variety of computational analysis methods, data standards, and public databases for the sharing of results.
The toponome is the spatial network code of proteins and other biomolecules in morphologically intact cells and tissues. It is mapped and decoded by imaging cycler microscopy (ICM) in situ able to co-map many thousand supermolecules in one sample. The term "toponome" is derived from the ancient Greek nouns "topos" and "nomos", and the term "toponomics" refers to the study of the toponome. It was introduced by Walter Schubert in 2003. It addresses the fact that the network of biomolecules in cells and tissues follows topological rules enabling coordinated actions. For example, the cell surface toponome provides the spatial protein interaction code for the execution of a cell movement, a "code of conduct". This is intrinsically dependent on the specific spatial arrangement of similar and dissimilar compositions of supermolecules with a specific spatial order along a cell surface membrane. This spatial order is periodically repeated when the cell tries to enter the exploratory state from the spherical state. This spatial toponome code is hierarchically organized with lead biomolecule(s), anti-colocated (absent) biomolecule(s) and wildcard molecules which are variably associated with the lead biomolecule(s). It has been shown that inhibition of lead molecule(s) in a surface membrane leads to disassembly of the corresponding biomolecular network and loss of function.
Toponomics is a discipline in systems biology, molecular cell biology, and histology concerning the study of the toponome of organisms. It is the field of study that purposes to decode the complete toponome in health and disease —which is the next big challenge in human biotechnology after having decoded the human genome.
FAST is a small, genetically-encoded, protein tag which allows for fluorescence reporting of proteins of interest. Unlike natural fluorescent proteins and derivates such as GFP or mCherry, FAST is not fluorescent by itself. It can bind selectively a fluorogenic chromophore derived from 4-hydroxybenzylidene rhodanine (HBR), which is itself non fluorescent unless bound. Once bound, the pair of molecules goes through a unique fluorogen activation mechanism based on two spectroscopic changes, increase of fluorescence quantum yield and absorption red shift, hence providing high labeling selectivity. The FAST-fluorogen reporting system can be used in fluorescence microscopy, flow cytometry and any other fluorometric method to explore the living world: biosensors, protein trafficking.
