The muscular system is an organ system consisting of skeletal, smooth, and cardiac muscle. It permits movement of the body, maintains posture, and circulates blood throughout the body. The muscular systems in vertebrates are controlled through the nervous system although some muscles can be completely autonomous. Together with the skeletal system in the human, it forms the musculoskeletal system, which is responsible for the movement of the body.
Myotonia is a symptom of a small handful of certain neuromuscular disorders characterized by delayed relaxation of the skeletal muscles after voluntary contraction or electrical stimulation, and the muscle shows an abnormal EMG.
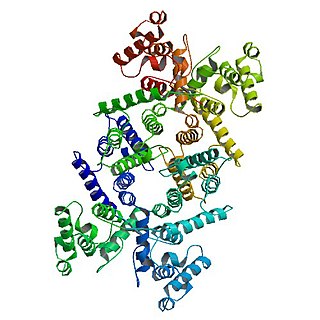
Dystrophin is a rod-shaped cytoplasmic protein, and a vital part of a protein complex that connects the cytoskeleton of a muscle fiber to the surrounding extracellular matrix through the cell membrane. This complex is variously known as the costamere or the dystrophin-associated protein complex (DAPC). Many muscle proteins, such as α-dystrobrevin, syncoilin, synemin, sarcoglycan, dystroglycan, and sarcospan, colocalize with dystrophin at the costamere. It has a molecular weight of 427 kDa
Hypotonia is a state of low muscle tone, often involving reduced muscle strength. Hypotonia is not a specific medical disorder, but a potential manifestation of many different diseases and disorders that affect motor nerve control by the brain or muscle strength. Hypotonia is a lack of resistance to passive movement, whereas muscle weakness results in impaired active movement. Central hypotonia originates from the central nervous system, while peripheral hypotonia is related to problems within the spinal cord, peripheral nerves and/or skeletal muscles. Severe hypotonia in infancy is commonly known as floppy baby syndrome. Recognizing hypotonia, even in early infancy, is usually relatively straightforward, but diagnosing the underlying cause can be difficult and often unsuccessful. The long-term effects of hypotonia on a child's development and later life depend primarily on the severity of the muscle weakness and the nature of the cause. Some disorders have a specific treatment but the principal treatment for most hypotonia of idiopathic or neurologic cause is physical therapy and/or occupational therapy for remediation.

Fukuyama congenital muscular dystrophy (FCMD) is a rare, autosomal recessive form of muscular dystrophy (weakness and breakdown of muscular tissue) mainly described in Japan but also identified in Turkish and Ashkenazi Jewish patients; fifteen cases were first described on 1960 by Dr. Yukio Fukuyama.
In medicine, myopathy is a disease of the muscle in which the muscle fibers do not function properly. Myopathy means muscle disease. This meaning implies that the primary defect is within the muscle, as opposed to the nerves or elsewhere.
Nemaline myopathy is a congenital, often hereditary neuromuscular disorder with many symptoms that can occur such as muscle weakness, hypoventilation, swallowing dysfunction, and impaired speech ability. The severity of these symptoms varies and can change throughout one's life to some extent. The prevalence is estimated at 1 in 50,000 live births. It is the most common non-dystrophic myopathy.

Mitochondrial myopathies are types of myopathies associated with mitochondrial disease. Adenosine triphosphate (ATP), the chemical used to provide energy for the cell, cannot be produced sufficiently by oxidative phosphorylation when the mitochondrion is either damaged or missing necessary enzymes or transport proteins. With ATP production deficient in mitochondria, there is an over-reliance on anaerobic glycolysis which leads to lactic acidosis either at rest or exercise-induced.

Dysferlin also known as dystrophy-associated fer-1-like protein is a protein that in humans is encoded by the DYSF gene. Dysferlin is linked with plasma membrane repair., stabilization of calcium signaling and the development of the T-tubule system of the muscle A defect in the DYSF gene, located on chromosome 2p12-14, results in several types of muscular dystrophy; including Miyoshi myopathy (MM), Limb-girdle muscular dystrophy type 2B (LGMD2B) and Distal Myopathy (DM). A reduction or absence of dysferlin, termed dysferlinopathy, usually becomes apparent in the third or fourth decade of life and is characterised by weakness and wasting of various voluntary skeletal muscles. Pathogenic mutations leading to dysferlinopathy can occur throughout the DYSF gene.

Centronuclear myopathies (CNM) are a group of congenital myopathies where cell nuclei are abnormally located in the center of muscle cells instead of their normal location at the periphery.
Congenital muscular dystrophies are autosomal recessively-inherited muscle diseases. They are a group of heterogeneous disorders characterized by muscle weakness which is present at birth and the different changes on muscle biopsy that ranges from myopathic to overtly dystrophic due to the age at which the biopsy takes place.

Distal myopathy is a group of rare genetic disorders that cause muscle damage and weakness, predominantly in the hands and/or feet. Mutation of many different genes can be causative. Many types involve dysferlin.
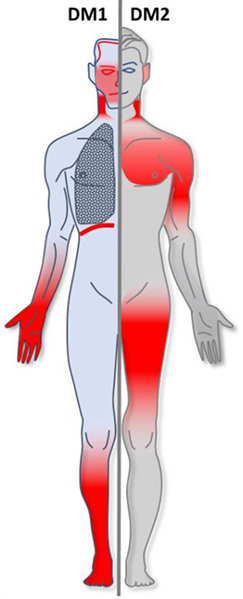
Myotonic dystrophy (DM) is a type of muscular dystrophy, a group of genetic disorders that cause progressive muscle loss and weakness. In DM, muscles are often unable to relax after contraction. Other manifestations may include cataracts, intellectual disability and heart conduction problems. In men, there may be early balding and infertility. While myotonic dystrophy can occur at any age, onset is typically in the 20s and 30s.
Congenital myopathy is a very broad term for any muscle disorder present at birth. This defect primarily affects skeletal muscle fibres and causes muscular weakness and/or hypotonia. Congenital myopathies account for one of the top neuromuscular disorders in the world today, comprising approximately 6 in 100,000 live births every year. As a whole, congenital myopathies can be broadly classified as follows:

Bethlem myopathy is predominantly an autosomal dominant myopathy, classified as a congenital form of limb-girdle muscular dystrophy. There are two types of Bethlem myopathy, based on which type of collagen is affected.
Ullrich congenital muscular dystrophy (UCMD) is a form of congenital muscular dystrophy. There are two forms: UCMD1 and UCMD2.
Collagen VI (ColVI) is a type of collagen primarily associated with the extracellular matrix of skeletal muscle. ColVI maintains regularity in muscle function and stabilizes the cell membrane. It is synthesized by a complex, multistep pathway that leads to the formation of a unique network of linked microfilaments located in the extracellular matrix (ECM). ColVI plays a vital role in numerous cell types, including chondrocytes, neurons, myocytes, fibroblasts, and cardiomyocytes. ColVI molecules are made up of three alpha chains: α1(VI), α2(VI), and α3(VI). It is encoded by 6 genes: COL6A1, COL6A2, COL6A3, COL6A4, COL6A5, and COL6A6. The chain lengths of α1(VI) and α2(VI) are about 1,000 amino acids. The chain length of α3(VI) is roughly a third larger than those of α1(VI) and α2(VI), and it consists of several spliced variants within the range of 2,500 to 3,100 amino acids.

X-linked myotubular myopathy (MTM) is a form of centronuclear myopathy (CNM) associated with mutations in the myotubularin 1 gene. It is found almost always in male infants. It is one of the severest congenital muscle diseases and is characterized by marked muscle weakness, hypotonia and feeding and breathing difficulties.
Acquired non-inflammatory myopathy (ANIM) is a neuromuscular disorder primarily affecting skeletal muscle, most commonly in the limbs of humans, resulting in a weakness or dysfunction in the muscle. A myopathy refers to a problem or abnormality with the myofibrils, which compose muscle tissue. In general, non-inflammatory myopathies are a grouping of muscular diseases not induced by an autoimmune-mediated inflammatory pathway. These muscular diseases usually arise from a pathology within the muscle tissue itself rather than the nerves innervating that tissue. ANIM has a wide spectrum of causes which include drugs and toxins, nutritional imbalances, acquired metabolic dysfunctions such as an acquired defect in protein structure, and infections.

Pseudohypertrophy, or false enlargement, is an increase in the size of an organ due to infiltration of a tissue not normally found in that organ. It is commonly applied to enlargement of a muscle due to infiltration of fat or connective tissue, famously in Duchenne muscular dystrophy. This is in contrast with typical muscle hypertrophy, in which the muscle tissue itself increases in size. Because pseudohypertrophy is not a result of increased muscle tissue, the muscles look bigger but are actually atrophied and thus weaker. Pseudohypertrophy is typically the result of a disease, which can be a disease of muscle or a disease of the nerve supplying the muscle.
