
In biology, a mutation is an alteration in the nucleic acid sequence of the genome of an organism, virus, or extrachromosomal DNA. Viral genomes contain either DNA or RNA. Mutations result from errors during DNA or viral replication, mitosis, or meiosis or other types of damage to DNA, which then may undergo error-prone repair, cause an error during other forms of repair, or cause an error during replication. Mutations may also result from insertion or deletion of segments of DNA due to mobile genetic elements.
Genetic drift, also known as random genetic drift, allelic drift or the Wright effect, is the change in the frequency of an existing gene variant (allele) in a population due to random chance.
Molecular evolution is the process of change in the sequence composition of cellular molecules such as DNA, RNA, and proteins across generations. The field of molecular evolution uses principles of evolutionary biology and population genetics to explain patterns in these changes. Major topics in molecular evolution concern the rates and impacts of single nucleotide changes, neutral evolution vs. natural selection, origins of new genes, the genetic nature of complex traits, the genetic basis of speciation, the evolution of development, and ways that evolutionary forces influence genomic and phenotypic changes.

The neutral theory of molecular evolution holds that most evolutionary changes occur at the molecular level, and most of the variation within and between species are due to random genetic drift of mutant alleles that are selectively neutral. The theory applies only for evolution at the molecular level, and is compatible with phenotypic evolution being shaped by natural selection as postulated by Charles Darwin.
Population genetics is a subfield of genetics that deals with genetic differences within and among populations, and is a part of evolutionary biology. Studies in this branch of biology examine such phenomena as adaptation, speciation, and population structure.
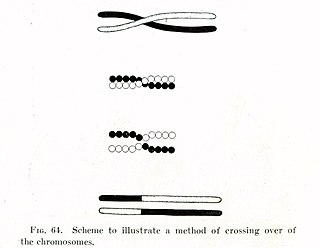
In evolutionary genetics, Muller's ratchet is a process which, in the absence of recombination, results in an accumulation of irreversible deleterious mutations. This happens because in the absence of recombination, and assuming reverse mutations are rare, offspring bear at least as much mutational load as their parents. Muller proposed this mechanism as one reason why sexual reproduction may be favored over asexual reproduction, as sexual organisms benefit from recombination and consequent elimination of deleterious mutations. The negative effect of accumulating irreversible deleterious mutations may not be prevalent in organisms which, while they reproduce asexually, also undergo other forms of recombination. This effect has also been observed in those regions of the genomes of sexual organisms that do not undergo recombination.

Motoo Kimura was a Japanese biologist best known for introducing the neutral theory of molecular evolution in 1968. He became one of the most influential theoretical population geneticists. He is remembered in genetics for his innovative use of diffusion equations to calculate the probability of fixation of beneficial, deleterious, or neutral alleles. Combining theoretical population genetics with molecular evolution data, he also developed the neutral theory of molecular evolution in which genetic drift is the main force changing allele frequencies. James F. Crow, himself a renowned population geneticist, considered Kimura to be one of the two greatest evolutionary geneticists, along with Gustave Malécot, after the great trio of the modern synthesis, Ronald Fisher, J. B. S. Haldane, and Sewall Wright.

Haldane's dilemma, also known as the waiting time problem, is a limit on the speed of beneficial evolution, calculated by J. B. S. Haldane in 1957. Before the invention of DNA sequencing technologies, it was not known how much polymorphism DNA harbored, although alloenzymes were beginning to make it clear that substantial polymorphism existed. This was puzzling because the amount of polymorphism known to exist seemed to exceed the theoretical limits that Haldane calculated, that is, the limits imposed if polymorphisms present in the population generally influence an organism's fitness. Motoo Kimura's landmark paper on neutral theory in 1968 built on Haldane's work to suggest that most molecular evolution is neutral, resolving the dilemma. Although neutral evolution remains the consensus theory among modern biologists, and thus Kimura's resolution of Haldane's dilemma is widely regarded as correct, some biologists argue that adaptive evolution explains a large fraction of substitutions in protein coding sequence, and they propose alternative solutions to Haldane's dilemma.
The effective population size (Ne) is size of an idealised population would experience the same rate of genetic drift or increase in inbreeding as in the real population. Idealised populations are based on unrealistic but convenient assumptions including random mating, simultaneous birth of each new generation, constant population size. For most quantities of interest and most real populations, Ne is smaller than the census population size N of a real population. The same population may have multiple effective population sizes for different properties of interest, including genetic drift and inbreeding.
Genetic load is the difference between the fitness of an average genotype in a population and the fitness of some reference genotype, which may be either the best present in a population, or may be the theoretically optimal genotype. The average individual taken from a population with a low genetic load will generally, when grown in the same conditions, have more surviving offspring than the average individual from a population with a high genetic load. Genetic load can also be seen as reduced fitness at the population level compared to what the population would have if all individuals had the reference high-fitness genotype. High genetic load may put a population in danger of extinction.
Mutation–selection balance is an equilibrium in the number of deleterious alleles in a population that occurs when the rate at which deleterious alleles are created by mutation equals the rate at which deleterious alleles are eliminated by selection. The majority of genetic mutations are neutral or deleterious; beneficial mutations are relatively rare. The resulting influx of deleterious mutations into a population over time is counteracted by negative selection, which acts to purge deleterious mutations. Setting aside other factors, the equilibrium number of deleterious alleles is then determined by a balance between the deleterious mutation rate and the rate at which selection purges those mutations.

Tomoko Ohta is a Japanese scientist and Professor Emeritus of the National Institute of Genetics. Ohta works on population genetics/molecular evolution and is known for developing the nearly neutral theory of evolution.
Coalescent theory is a model of how alleles sampled from a population may have originated from a common ancestor. In the simplest case, coalescent theory assumes no recombination, no natural selection, and no gene flow or population structure, meaning that each variant is equally likely to have been passed from one generation to the next. The model looks backward in time, merging alleles into a single ancestral copy according to a random process in coalescence events. Under this model, the expected time between successive coalescence events increases almost exponentially back in time. Variance in the model comes from both the random passing of alleles from one generation to the next, and the random occurrence of mutations in these alleles.
Genetic hitchhiking, also called genetic draft or the hitchhiking effect, is when an allele changes frequency not because it itself is under natural selection, but because it is near another gene that is undergoing a selective sweep and that is on the same DNA chain. When one gene goes through a selective sweep, any other nearby polymorphisms that are in linkage disequilibrium will tend to change their allele frequencies too. Selective sweeps happen when newly appeared mutations are advantageous and increase in frequency. Neutral or even slightly deleterious alleles that happen to be close by on the chromosome 'hitchhike' along with the sweep. In contrast, effects on a neutral locus due to linkage disequilibrium with newly appeared deleterious mutations are called background selection. Both genetic hitchhiking and background selection are stochastic (random) evolutionary forces, like genetic drift.

The Neutral Theory of Molecular Evolution is an influential monograph written in 1983 by Japanese evolutionary biologist Motoo Kimura. While the neutral theory of molecular evolution existed since his article in 1968, Kimura felt the need to write a monograph with up-to-date information and evidences showing the importance of his theory in evolution.
Neutral mutations are changes in DNA sequence that are neither beneficial nor detrimental to the ability of an organism to survive and reproduce. In population genetics, mutations in which natural selection does not affect the spread of the mutation in a species are termed neutral mutations. Neutral mutations that are inheritable and not linked to any genes under selection will be lost or will replace all other alleles of the gene. That loss or fixation of the gene proceeds based on random sampling known as genetic drift. A neutral mutation that is in linkage disequilibrium with other alleles that are under selection may proceed to loss or fixation via genetic hitchhiking and/or background selection.
In population genetics, fixation is the change in a gene pool from a situation where there exists at least two variants of a particular gene (allele) in a given population to a situation where only one of the alleles remains. That is, the allele becomes fixed. In the absence of mutation or heterozygote advantage, any allele must eventually be lost completely from the population or fixed. Whether a gene will ultimately be lost or fixed is dependent on selection coefficients and chance fluctuations in allelic proportions. Fixation can refer to a gene in general or particular nucleotide position in the DNA chain (locus).
The history of molecular evolution starts in the early 20th century with "comparative biochemistry", but the field of molecular evolution came into its own in the 1960s and 1970s, following the rise of molecular biology. The advent of protein sequencing allowed molecular biologists to create phylogenies based on sequence comparison, and to use the differences between homologous sequences as a molecular clock to estimate the time since the last common ancestor. In the late 1960s, the neutral theory of molecular evolution provided a theoretical basis for the molecular clock, though both the clock and the neutral theory were controversial, since most evolutionary biologists held strongly to panselectionism, with natural selection as the only important cause of evolutionary change. After the 1970s, nucleic acid sequencing allowed molecular evolution to reach beyond proteins to highly conserved ribosomal RNA sequences, the foundation of a reconceptualization of the early history of life.
A nonsynonymous substitution is a nucleotide mutation that alters the amino acid sequence of a protein. Nonsynonymous substitutions differ from synonymous substitutions, which do not alter amino acid sequences and are (sometimes) silent mutations. As nonsynonymous substitutions result in a biological change in the organism, they are subject to natural selection.
The McDonald–Kreitman test is a statistical test often used by evolutionary and population biologists to detect and measure the amount of adaptive evolution within a species by determining whether adaptive evolution has occurred, and the proportion of substitutions that resulted from positive selection. To do this, the McDonald–Kreitman test compares the amount of variation within a species (polymorphism) to the divergence between species (substitutions) at two types of sites, neutral and nonneutral. A substitution refers to a nucleotide that is fixed within one species, but a different nucleotide is fixed within a second species at the same base pair of homologous DNA sequences. A site is nonneutral if it is either advantageous or deleterious. The two types of sites can be either synonymous or nonsynonymous within a protein-coding region. In a protein-coding sequence of DNA, a site is synonymous if a point mutation at that site would not change the amino acid, also known as a silent mutation. Because the mutation did not result in a change in the amino acid that was originally coded for by the protein-coding sequence, the phenotype, or the observable trait, of the organism is generally unchanged by the silent mutation. A site in a protein-coding sequence of DNA is nonsynonymous if a point mutation at that site results in a change in the amino acid, resulting in a change in the organism's phenotype. Typically, silent mutations in protein-coding regions are used as the "control" in the McDonald–Kreitman test.














