





Partner and localizer of BRCA2, also known as PALB2 or FANCN, is a protein which in humans is encoded by the PALB2 gene. [5] [6] [7]
Partner and localizer of BRCA2, also known as PALB2 or FANCN, is a protein which in humans is encoded by the PALB2 gene. [5] [6] [7]
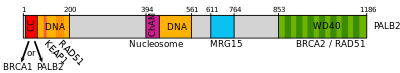

This gene encodes a protein that functions in genome maintenance (double strand break repair). This protein binds to and colocalizes with the breast cancer 2 early onset protein (BRCA2) in nuclear foci and likely permits the stable intranuclear localization and accumulation of BRCA2. [5] PALB2 binds the single strand DNA and directly interacts with the recombinase RAD51 to stimulate strand invasion, a vital step of homologous recombination, [15] PALB2 can function synergistically with a BRCA2 chimera (termed piccolo, or piBRCA2) to further promote strand invasion. [15]
Variants in the PALB2 gene are associated with an increased risk of developing breast cancer [16] of magnitude similar to that associated with BRCA2 mutations [17] and PALB2-deficient cells are sensitive to PARP inhibitors. [15]
PALB2 was recently identified as a susceptibility gene for familial pancreatic cancer by scientists at the Sol Goldman Pancreatic Cancer Research Center at Johns Hopkins. This has paved for the way for developing a new gene test for families where pancreatic cancer occurs in multiple family members. [18] Tests for PALB2 have been developed by Ambry Genetics [19] and Myriad Genetics [20] that are now available.
Prophylactic mastectomy should be considered for women that had breast cancer and a PALB2 mutation. [21] [22]
Biallelic mutations in PALB2 (also known as FANCN), similar to biallelic BRCA2 mutations, cause Fanconi anemia. [7]
Mutations in this gene have been associated with an increased risk of ovarian, breast and pancreatic cancer. [23]
PALB2 mutant male mice have reduced fertility. [24] This reduced fertility appears to be due to germ cell attrition resulting from a combination of unrepaired DNA breaks during meiosis and defective synapsis of the X and Y chromosomes. The function of homologous recombination during meiosis appears to be repair of DNA damages, particularly double-strand breaks (also see Origin and function of meiosis).[ citation needed ] The PALB2-BRCA1 interaction is likely important for repairing such damages during male meiosis.

Breast cancer type 1 susceptibility protein is a protein that in humans is encoded by the BRCA1 gene. Orthologs are common in other vertebrate species, whereas invertebrate genomes may encode a more distantly related gene. BRCA1 is a human tumor suppressor gene and is responsible for repairing DNA.

Fanconi anaemia (FA) is a rare genetic disease resulting in impaired response to DNA damage. Although it is a very rare disorder, study of this and other bone marrow failure syndromes has improved scientific understanding of the mechanisms of normal bone marrow function and development of cancer. Among those affected, the majority develop cancer, most often acute myelogenous leukemia (AML), and 90% develop aplastic anemia by age 40. About 60–75% have congenital defects, commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and developmental disabilities. Around 75% have some form of endocrine problem, with varying degrees of severity.

BRCA2 and BRCA2 are a human gene and its protein product, respectively. The official symbol and the official name are maintained by the HUGO Gene Nomenclature Committee. One alternative symbol, FANCD1, recognizes its association with the FANC protein complex. Orthologs, styled Brca2 and Brca2, are common in other vertebrate species. BRCA2 is a human tumor suppressor gene, found in all humans; its protein, also called by the synonym breast cancer type 2 susceptibility protein, is responsible for repairing DNA.

ATM serine/threonine kinase or Ataxia-telangiectasia mutated, symbol ATM, is a serine/threonine protein kinase that is recruited and activated by DNA double-strand breaks. It phosphorylates several key proteins that initiate activation of the DNA damage checkpoint, leading to cell cycle arrest, DNA repair or apoptosis. Several of these targets, including p53, CHK2, BRCA1, NBS1 and H2AX are tumor suppressors.
DNA repair protein RAD51 homolog 1 is a protein encoded by the gene RAD51. The enzyme encoded by this gene is a member of the RAD51 protein family which assists in repair of DNA double strand breaks. RAD51 family members are homologous to the bacterial RecA, Archaeal RadA and yeast Rad51. The protein is highly conserved in most eukaryotes, from yeast to humans.

CHEK2 is a tumor suppressor gene that encodes the protein CHK2, a serine-threonine kinase. CHK2 is involved in DNA repair, cell cycle arrest or apoptosis in response to DNA damage. Mutations to the CHEK2 gene have been linked to a wide range of cancers.

Hereditary breast–ovarian cancer syndromes (HBOC) are cancer syndromes that produce higher than normal levels of breast cancer, ovarian cancer and additional cancers in genetically related families. It accounts for 90% of the hereditary cancers. The hereditary factors may be proven or suspected to cause the pattern of breast and ovarian cancer occurrences in the family. The name HBOC may be misleading because it implies that this genetic susceptibility to cancer is mainly in women. In reality, both sexes have the same rates of gene mutations and HBOC can predispose to other cancers including prostate cancer and pancreatic cancer. For this reason, the term "King syndrome" has recently come into use. The new name references Mary-Claire King who identified the genes BRCA1 and BRCA2.
Lethal alleles are alleles that cause the death of the organism that carries them. They are usually a result of mutations in genes that are essential for growth or development. Lethal alleles may be recessive, dominant, or conditional depending on the gene or genes involved.

Double-strand break repair protein MRE11 is an enzyme that in humans is encoded by the MRE11 gene. The gene has been designated MRE11A to distinguish it from the pseudogene MRE11B that is nowadays named MRE11P1.

Fanconi anaemia, complementation group A, also known as FAA, FACA and FANCA, is a protein which in humans is encoded by the FANCA gene. It belongs to the Fanconi anaemia complementation group (FANC) family of genes of which 12 complementation groups are currently recognized and is hypothesised to operate as a post-replication repair or a cell cycle checkpoint. FANCA proteins are involved in inter-strand DNA cross-link repair and in the maintenance of normal chromosome stability that regulates the differentiation of haematopoietic stem cells into mature blood cells.

Fanconi anemia group D2 protein is a protein that in humans is encoded by the FANCD2 gene. The Fanconi anemia complementation group (FANC) currently includes FANCA, FANCB, FANCC, FANCD1, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN and FANCO.

Fanconi anemia group G protein is a protein that in humans is encoded by the FANCG gene.

Fanconi anemia group J protein is a protein that in humans is encoded by the BRCA1-interacting protein 1 (BRIP1) gene.

Fanconi anemia group F protein is a protein that in humans is encoded by the FANCF gene.
Fanconi anemia, complementation group E protein is a protein that in humans is encoded by the FANCE gene. The Fanconi anemia complementation group (FANC) currently includes FANCA, FANCB, FANCC, FANCD1, FANCD2, FANCE, FANCF, FANCG, and FANCL. Fanconi anemia is a genetically heterogeneous recessive disorder characterized by cytogenetic instability, hypersensitivity to DNA cross-linking agents, increased chromosomal breakage, and defective DNA repair. The members of the Fanconi anemia complementation group do not share sequence similarity; they are related by their assembly into a common nuclear protein complex. This gene encodes the protein for complementation groufcrp E.

E3 ubiquitin-protein ligase FANCL is an enzyme that in humans is encoded by the FANCL gene.

Fanconi anemia group B protein is a protein that in humans is encoded by the FANCB gene.
Fanconi anemia, complementation group M, also known as FANCM is a human gene. It is an emerging target in cancer therapy, in particular cancers with specific genetic deficiencies.
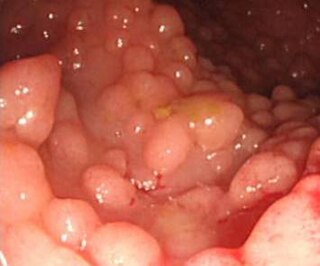
A cancer syndrome, or family cancer syndrome, is a genetic disorder in which inherited genetic mutations in one or more genes predispose the affected individuals to the development of cancers and may also cause the early onset of these cancers. Cancer syndromes often show not only a high lifetime risk of developing cancer, but also the development of multiple independent primary tumors.
Bing Xia is a Chinese American scientist and professor at the Rutgers Cancer Institute of New Jersey, where he directs the Xia Laboratory. He is best known for his discovery of the PALB2 tumor suppressor gene.