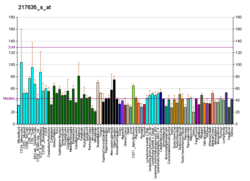
A DNA polymerase is a member of a family of enzymes that catalyze the synthesis of DNA molecules from nucleoside triphosphates, the molecular precursors of DNA. These enzymes are essential for DNA replication and usually work in groups to create two identical DNA duplexes from a single original DNA duplex. During this process, DNA polymerase "reads" the existing DNA strands to create two new strands that match the existing ones. These enzymes catalyze the chemical reaction

Human mitochondrial genetics is the study of the genetics of human mitochondrial DNA. The human mitochondrial genome is the entirety of hereditary information contained in human mitochondria. Mitochondria are small structures in cells that generate energy for the cell to use, and are hence referred to as the "powerhouses" of the cell.
T7 DNA helicase (gp4) is a hexameric motor protein encoded by T7 phages that uses energy from dTTP hydrolysis to process unidirectionally along single stranded DNA, separating (helicase) the two strands as it progresses. It is also a primase, making short stretches of RNA that initiates DNA synthesis. It forms a complex with T7 DNA polymerase. Its homologs are found in mitochondria and chloroplasts.
Chronic progressive external ophthalmoplegia (CPEO) is a type of eye disorder characterized by slowly progressive inability to move the eyes and eyebrows. It is often the only feature of mitochondrial disease, in which case the term CPEO may be given as the diagnosis. In other people suffering from mitochondrial disease, CPEO occurs as part of a syndrome involving more than one part of the body, such as Kearns–Sayre syndrome. Occasionally CPEO may be caused by conditions other than mitochondrial diseases.

DNA polymerase delta catalytic subunit(DPOD1) is an enzyme that is encoded in the human by the POLD1 gene, in the DNA polymerase delta complex. DPOD1 is responsible for synthesizing the lagging strand of DNA, and has also been implicated in some activities at the leading strand. The DPOD1 subunit encodes both DNA polymerizing and exonuclease domains, which provide the protein an important second function in proofreading to ensure replication accuracy during DNA synthesis, and in a number of types of replication-linked DNA repair following DNA damage.

Twinkle protein also known as twinkle mtDNA helicase is a mitochondrial protein that in humans is encoded by the TWNK gene located in the long arm of chromosome 10 (10q24.31).

Ribonucleotide-diphosphate reductase subunit M2 B is an enzyme that in humans is encoded by the RRM2B gene. The gene encoding the RRM2B protein is located on chromosome 8, at position 8q23.1. The gene and its products are also known by designations MTDPS8A, MTDPS8B, and p53R2.
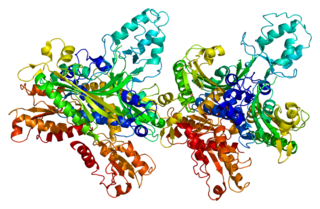
DNA polymerase subunit gamma-2, mitochondrial is a protein that in humans is encoded by the POLG2 gene. The POLG2 gene encodes a 55 kDa accessory subunit protein that imparts high processivity and salt tolerance to the catalytic subunit of DNA polymerase gamma, encoded by the POLG gene. Mutations in this gene result in autosomal dominant progressive external ophthalmoplegia with mitochondrial DNA deletions.

NADH dehydrogenase [ubiquinone] flavoprotein 1, mitochondrial (NDUFV1) is an enzyme that in humans is encoded by the NDUFV1 gene. The NDUFV1 gene encodes the 51-kD subunit of complex I of the mitochondrial respiratory chain. Defects in complex I are a common cause of mitochondrial dysfunction. Mitochondrial complex I deficiency is linked to myopathies, encephalomyopathies, and neurodegenerative disorders such as Parkinson's disease and Leigh syndrome.
NADH dehydrogenase [ubiquinone] iron-sulfur protein 8, mitochondrial also known as NADH-ubiquinone oxidoreductase 23 kDa subunit, Complex I-23kD (CI-23kD), or TYKY subunit is an enzyme that in humans is encoded by the NDUFS8 gene. The NDUFS8 protein is a subunit of NADH dehydrogenase (ubiquinone) also known as Complex I, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Mutations in this gene have been associated with Leigh syndrome.
NADH dehydrogenase [ubiquinone] iron-sulfur protein 7, mitochondrial, also knowns as NADH-ubiquinone oxidoreductase 20 kDa subunit, Complex I-20kD (CI-20kD), or PSST subunit is an enzyme that in humans is encoded by the NDUFS7 gene. The NDUFS7 protein is a subunit of NADH dehydrogenase (ubiquinone) also known as Complex I, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain.

DNA primase large subunit is an enzyme that in humans is encoded by the PRIM2 gene.
Mitochondrially encoded tRNA asparagine also known as MT-TN is a transfer RNA which in humans is encoded by the mitochondrial MT-TN gene.
Mitochondrially encoded tRNA tyrosine, also known as MT-TY, is a transfer RNA which in humans is encoded by the mitochondrial MT-TY gene.
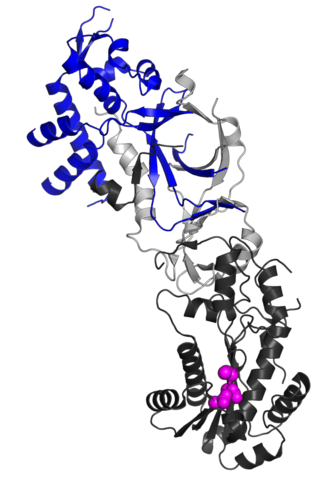
Ribonuclease H2, subunit B is a protein in humans is encoded by the RNASEH2B gene. RNase H2 is composed of a single catalytic subunit (A) and two non-catalytic subunits, and degrades the RNA of RNA:DNA hybrids. The non-catalytic B subunit of RNase H2 is thought to play a role in DNA replication.

Mitochondrial DNA depletion syndrome, or Alper's disease, is any of a group of autosomal recessive disorders that cause a significant drop in mitochondrial DNA in affected tissues. Symptoms can be any combination of myopathic, hepatopathic, or encephalomyopathic. These syndromes affect tissue in the muscle, liver, or both the muscle and brain, respectively. The condition is typically fatal in infancy and early childhood, though some have survived to their teenage years with the myopathic variant and some have survived into adulthood with the SUCLA2 encephalomyopathic variant. There is currently no curative treatment for any form of MDDS, though some preliminary treatments have shown a reduction in symptoms.

PrimPol is a protein encoded by the PRIMPOL gene in humans. PrimPol is a eukaryotic protein with both DNA polymerase and DNA Primase activities involved in translesion DNA synthesis. It is the first eukaryotic protein to be identified with priming activity using deoxyribonucleotides. It is also the first protein identified in the mitochondria to have translesion DNA synthesis activities.

DNA primase small subunit is an enzyme that in humans is encoded by the PRIM1 gene.

DNA polymerase alpha catalytic subunit is an enzyme that in humans is encoded by the POLA1 gene.