Related Research Articles
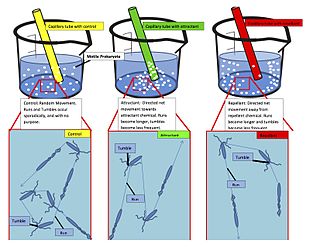
Chemotaxis is the movement of an organism or entity in response to a chemical stimulus. Somatic cells, bacteria, and other single-cell or multicellular organisms direct their movements according to certain chemicals in their environment. This is important for bacteria to find food by swimming toward the highest concentration of food molecules, or to flee from poisons. In multicellular organisms, chemotaxis is critical to early development and development as well as in normal function and health. In addition, it has been recognized that mechanisms that allow chemotaxis in animals can be subverted during cancer metastasis. The aberrant chemotaxis of leukocytes and lymphocytes also contribute to inflammatory diseases such as atherosclerosis, asthma, and arthritis. Sub-cellular components, such as the polarity patch generated by mating yeast, may also display chemotactic behavior.

Proteins are large biomolecules and macromolecules that comprise one or more long chains of amino acid residues. Proteins perform a vast array of functions within organisms, including catalysing metabolic reactions, DNA replication, responding to stimuli, providing structure to cells and organisms, and transporting molecules from one location to another. Proteins differ from one another primarily in their sequence of amino acids, which is dictated by the nucleotide sequence of their genes, and which usually results in protein folding into a specific 3D structure that determines its activity.

Protein folding is the physical process by which a protein chain is translated to its native three-dimensional structure, typically a "folded" conformation by which the protein becomes biologically functional. Via an expeditious and reproducible process, a polypeptide folds into its characteristic three-dimensional structure from a random coil. Each protein exists first as an unfolded polypeptide or random coil after being translated from a sequence of mRNA to a linear chain of amino acids. At this stage the polypeptide lacks any stable (long-lasting) three-dimensional structure. As the polypeptide chain is being synthesized by a ribosome, the linear chain begins to fold into its three-dimensional structure.

Peripheral membrane proteins, or extrinsic membrane proteins, are membrane proteins that adhere only temporarily to the biological membrane with which they are associated. These proteins attach to integral membrane proteins, or penetrate the peripheral regions of the lipid bilayer. The regulatory protein subunits of many ion channels and transmembrane receptors, for example, may be defined as peripheral membrane proteins. In contrast to integral membrane proteins, peripheral membrane proteins tend to collect in the water-soluble component, or fraction, of all the proteins extracted during a protein purification procedure. Proteins with GPI anchors are an exception to this rule and can have purification properties similar to those of integral membrane proteins.

Aspartate carbamoyltransferase catalyzes the first step in the pyrimidine biosynthetic pathway.
PEP group translocation, also known as the phosphotransferase system or PTS, is a distinct method used by bacteria for sugar uptake where the source of energy is from phosphoenolpyruvate (PEP). It is known to be a multicomponent system that always involves enzymes of the plasma membrane and those in the cytoplasm.
A tetrameric protein is a protein with a quaternary structure of four subunits (tetrameric). Homotetramers have four identical subunits, and heterotetramers are complexes of different subunits. A tetramer can be assembled as dimer of dimers with two homodimer subunits, or two heterodimer subunits.

The Matrix-2 (M2) protein is a proton-selective viroporin, integral in the viral envelope of the influenza A virus. The channel itself is a homotetramer, where the units are helices stabilized by two disulfide bonds, and is activated by low pH. The M2 protein is encoded on the seventh RNA segment together with the M1 protein. Proton conductance by the M2 protein in influenza A is essential for viral replication.
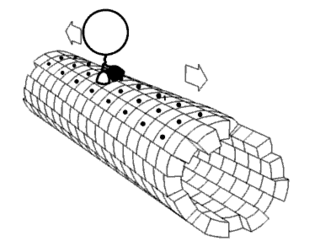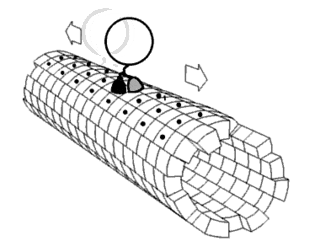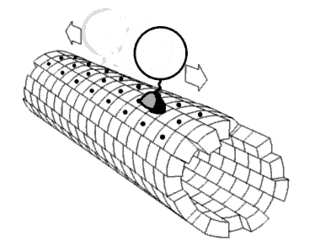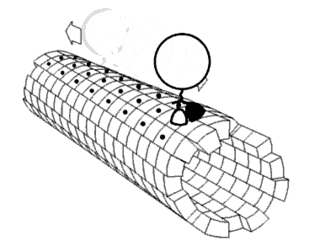
In biochemistry, a conformational change is a change in the shape of a macromolecule, often induced by environmental factors.

Tissue transglutaminase is a 78-kDa, calcium-dependent enzyme of the protein-glutamine γ-glutamyltransferases family. Like other transglutaminases, it crosslinks proteins between an ε-amino group of a lysine residue and a γ-carboxamide group of glutamine residue, creating an inter- or intramolecular bond that is highly resistant to proteolysis. Aside from its crosslinking function, tTG catalyzes other types of reactions including deamidation, GTP-binding/hydrolyzing, and isopeptidase activities. Unlike other members of the transglutaminase family, tTG can be found both in the intracellular and the extracellular spaces of various types of tissues and is found in many different organs including the heart, the liver, and the small intestine. Intracellular tTG is abundant in the cytosol but smaller amounts can also be found in the nucleus and the mitochondria. Intracellular tTG is thought to play an important role in apoptosis. In the extracellular space, tTG binds to proteins of the extracellular matrix (ECM), binding particularly tightly to fibronectin. Extracellular tTG has been linked to cell adhesion, ECM stabilization, wound healing, receptor signaling, cellular proliferation, and cellular motility.

The residual dipolar coupling between two spins in a molecule occurs if the molecules in solution exhibit a partial alignment leading to an incomplete averaging of spatially anisotropic dipolar couplings.
Cell-penetrating peptides (CPPs) are short peptides that facilitate cellular intake and uptake of molecules ranging from nanosize particles to small chemical compounds to large fragments of DNA. The "cargo" is associated with the peptides either through chemical linkage via covalent bonds or through non-covalent interactions.
Vitamin K-dependent carboxylation/gamma-carboxyglutamic (GLA) domain is a protein domain that contains post-translational modifications of many glutamate residues by vitamin K-dependent carboxylation to form γ-carboxyglutamate (Gla). Proteins with this domain are known informally as Gla proteins. The Gla residues are responsible for the high-affinity binding of calcium ions.

Nucleoporin 214 (Nup2014) is a protein that in humans is encoded by the NUP214 gene.

Chloride intracellular channel protein 1 is a protein that in humans is encoded by the CLIC1 gene.

The phenomenon of macromolecular crowding alters the properties of molecules in a solution when high concentrations of macromolecules such as proteins are present. Such conditions occur routinely in living cells; for instance, the cytosol of Escherichia coli contains about 300–400 mg/ml of macromolecules. Crowding occurs since these high concentrations of macromolecules reduce the volume of solvent available for other molecules in the solution, which has the result of increasing their effective concentrations. Crowding can promote formation of a biomolecular condensate by colloidal phase separation.
Experimental approaches of determining the structure of nucleic acids, such as RNA and DNA, can be largely classified into biophysical and biochemical methods. Biophysical methods use the fundamental physical properties of molecules for structure determination, including X-ray crystallography, NMR and cryo-EM. Biochemical methods exploit the chemical properties of nucleic acids using specific reagents and conditions to assay the structure of nucleic acids. Such methods may involve chemical probing with specific reagents, or rely on native or analogue chemistry. Different experimental approaches have unique merits and are suitable for different experimental purposes.
Sortase refers to a group of prokaryotic enzymes that modify surface proteins by recognizing and cleaving a carboxyl-terminal sorting signal. For most substrates of sortase enzymes, the recognition signal consists of the motif LPXTG (Leu-Pro-any-Thr-Gly), then a highly hydrophobic transmembrane sequence, followed by a cluster of basic residues such as arginine. Cleavage occurs between the Thr and Gly, with transient attachment through the Thr residue to the active site Cys residue, followed by transpeptidation that attaches the protein covalently to cell wall components. Sortases occur in almost all Gram-positive bacteria and the occasional Gram-negative bacterium or Archaea, where cell wall LPXTG-mediated decoration has not been reported. Although sortase A, the "housekeeping" sortase, typically acts on many protein targets, other forms of sortase recognize variant forms of the cleavage motif, or catalyze the assembly of pilins into pili.
Adenovirus early region 1A (E1A) is a gene expressed during adenovirus replication to produce a variety of E1A proteins. It is expressed during the early phase of the viral life span.

G. Marius Clore MAE, FRSC, FRS is a British-born, Anglo-American molecular biophysicist and structural biologist. He was born in London, U.K. and is a dual US/U.K. Citizen. He is a Member of the National Academy of Sciences, a Fellow of the Royal Society, a NIH Distinguished Investigator, and the Chief of the Molecular and Structural Biophysics Section in the Laboratory of Chemical Physics of the National Institute of Diabetes and Digestive and Kidney Diseases at the U.S. National Institutes of Health. He is known for his foundational work in three-dimensional protein and nucleic acid structure determination by biomolecular NMR spectroscopy, for advancing experimental approaches to the study of large macromolecules and their complexes by NMR, and for developing NMR-based methods to study rare conformational states in protein-nucleic acid and protein-protein recognition. Clore's discovery of previously undetectable, functionally significant, rare transient states of macromolecules has yielded fundamental new insights into the mechanisms of important biological processes, and in particular the significance of weak interactions and the mechanisms whereby the opposing constraints of speed and specificity are optimized. Further, Clore's work opens up a new era of pharmacology and drug design as it is now possible to target structures and conformations that have been heretofore unseen.
References
- ↑ Cohen, Rachel D.; Pielak, Gary J. (2016). "Electrostatic Contributions to Protein Quinary Structure". Journal of the American Chemical Society . 138 (40): 13139–13142. doi:10.1021/jacs.6b07323. PMID 27676610.
- ↑ Edelstein, S. J. (October 1980). "Patterns in the quinary structures of proteins. Plasticity and inequivalence of individual molecules in helical arrays of sickle cell hemoglobin and tubulin". Biophysical Journal. 32 (1): 347–360. Bibcode:1980BpJ....32..347E. doi:10.1016/S0006-3495(80)84961-7. PMC 1327314 . PMID 7248453.
- ↑ "Probing Protein Quinary Interactions by in-cell NMR". ResearchGate. Retrieved 2019-09-02.
- ↑ Shekhtman, Alexander; Burz, David S.; DeMott, Christopher; Breindel, Leonard (2018). "Real-Time In-Cell Nuclear Magnetic Resonance: Ribosome-Targeted Antibiotics Modulate Quinary Protein Interactions". Biochemistry. U.S.: United States Department of Agriculture. 57 (5): 540–546. doi:10.1021/acs.biochem.7b00938. PMC 5801172 . PMID 29266932 . Retrieved 2019-09-02.
- ↑ Danielsson, J.; Oliveberg, M. (2017). "Comparing protein behaviour in vitro and in vivo, what does the data really tell us?". Current Opinion in Structural Biology. 42: 129–135. doi:10.1016/j.sbi.2017.01.002. PMID 28126529.
- ↑ Jacek T. Mika; Bert Poolman (2011). "Macromolecule diffusion and confinement in prokaryotic cells". Current Opinion in Biotechnology. 22 (1): 117–126. doi:10.1016/j.copbio.2010.09.009. PMID 20952181.
- 1 2 McConkey, E. H. (1989). "Molecular evolution, intracellular organization, and the quinary structure of proteins". Proceedings of the National Academy of Sciences of the United States of America. 79 (10): 3236–3240. doi: 10.1073/pnas.79.10.3236 . PMC 346390 . PMID 6954476.
- ↑ Wlodarski, T.; Zagrovic, B. (2009). "Conformational selection and induced fit mechanism underlie specificity in noncovalent interactions with ubiquitin". Proceedings of the National Academy of Sciences of the United States of America. 106 (46): 3236–3240. Bibcode:2009PNAS..10619346W. doi: 10.1073/pnas.0906966106 . PMC 2780739 . PMID 19887638.
- ↑ Schreiber, G.; Fersht, A. R. (1996). "Rapid, electrostatically assisted association of proteins". Nature Structural Biology. 3 (5): 427–431. doi:10.1038/nsb0596-427. PMID 8612072. S2CID 25318867.
- ↑ Deeds, E. J.; Ashenberg, O.; Shakhnovich, E. I. (2006). "From The Cover: A simple physical model for scaling in protein-protein interaction networks". Proceedings of the National Academy of Sciences of the United States of America. 103 (2): 311–316. arXiv: q-bio/0509001 . Bibcode:2006PNAS..103..311D. doi: 10.1073/pnas.0509715102 . PMC 1326177 . PMID 16384916.
- ↑ Jian-Rong Yang; Ben-Yang Liao; Shi-Mei Zhuang; Jianzhi Zhang (2012). "Protein misinteraction avoidance causes highly expressed proteins to evolve slowly". Proceedings of the National Academy of Sciences of the United States of America. 109 (14): E831–E840. doi: 10.1073/pnas.1117408109 . PMC 3325723 . PMID 22416125.
- ↑ Wirth, A. J.; Gruebele, M. (2013). "Quinary protein structure and the consequences of crowding in living cells: Leaving the test-tube behind". BioEssays. 35 (11): 984–993. doi:10.1002/bies.201300080. PMID 23943406. S2CID 33478753.
- ↑ Peter B. Crowley; Elysian Chow; Tatiana Papkovskaia (2011). "Protein Interactions in the Escherichia coli Cytosol: An Impediment to In‐Cell NMR Spectroscopy". ChemBioChem. 12 (7): 1043–1048. doi:10.1002/cbic.201100063. PMID 21448871. S2CID 44250541.
- ↑ Xin Mu; Seongil Choi; Lisa Lang; David Mowray; Nikolay V. Dokholyan; Jens Danielsson; Mikael Oliveberg (2017). "Physicochemical code for quinary protein interactions in Escherichia coli". Proceedings of the National Academy of Sciences of the United States of America. 114 (23): E4556–E4563. Bibcode:2017PNAS..114E4556M. doi: 10.1073/pnas.1621227114 . PMC 5468600 . PMID 28536196.