The Baeyer–Villiger oxidation is an organic reaction that forms an ester from a ketone or a lactone from a cyclic ketone, using peroxyacids or peroxides as the oxidant. The reaction is named after Adolf von Baeyer and Victor Villiger who first reported the reaction in 1899.

In organic chemistry, transannular strain is the unfavorable interactions of ring substituents on non-adjacent carbons. These interactions, called transannular interactions, arise from a lack of space in the interior of the ring, which forces substituents into conflict with one another. In medium-sized cycloalkanes, which have between 8 and 11 carbons constituting the ring, transannular strain can be a major source of the overall strain, especially in some conformations, to which there is also contribution from large-angle strain and Pitzer strain. In larger rings, transannular strain drops off until the ring is sufficiently large that it can adopt conformations devoid of any negative interactions.
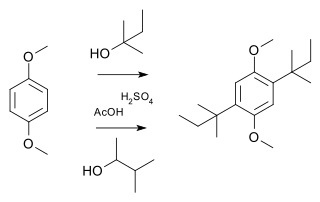
A Wagner–Meerwein rearrangement is a class of carbocation 1,2-rearrangement reactions in which a hydrogen, alkyl or aryl group migrates from one carbon to a neighboring carbon. They can be described as cationic [1,2]-sigmatropic rearrangements, proceeding suprafacially and with stereochemical retention. As such, a Wagner–Meerwein shift is a thermally allowed pericyclic process with the Woodward-Hoffmann symbol [ω0s + σ2s]. They are usually facile, and in many cases, they can take place at temperatures as low as –120 °C. The reaction is named after the Russian chemist Yegor Yegorovich Vagner; he had German origin and published in German journals as Georg Wagner; and Hans Meerwein.
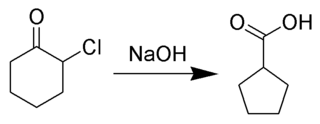
The Favorskii rearrangement is principally a rearrangement of cyclopropanones and α-halo ketones that leads to carboxylic acid derivatives. In the case of cyclic α-halo ketones, the Favorskii rearrangement constitutes a ring contraction. This rearrangement takes place in the presence of a base, sometimes hydroxide, to yield a carboxylic acid, but usually either an alkoxide base or an amine to yield an ester or an amide, respectively. α,α'-Dihaloketones eliminate HX under the reaction conditions to give α,β-unsaturated carbonyl compounds.

The Dakin oxidation (or Dakin reaction) is an organic redox reaction in which an ortho- or para-hydroxylated phenyl aldehyde (2-hydroxybenzaldehyde or 4-hydroxybenzaldehyde) or ketone reacts with hydrogen peroxide (H2O2) in base to form a benzenediol and a carboxylate. Overall, the carbonyl group is oxidised, whereas the H2O2 is reduced.

The pinacol–pinacolone rearrangement is a method for converting a 1,2-diol to a carbonyl compound in organic chemistry. The 1,2-rearrangement takes place under acidic conditions. The name of the rearrangement reaction comes from the rearrangement of pinacol to pinacolone.

A cyclic compound is a term for a compound in the field of chemistry in which one or more series of atoms in the compound is connected to form a ring. Rings may vary in size from three to many atoms, and include examples where all the atoms are carbon, none of the atoms are carbon, or where both carbon and non-carbon atoms are present. Depending on the ring size, the bond order of the individual links between ring atoms, and their arrangements within the rings, carbocyclic and heterocyclic compounds may be aromatic or non-aromatic; in the latter case, they may vary from being fully saturated to having varying numbers of multiple bonds between the ring atoms. Because of the tremendous diversity allowed, in combination, by the valences of common atoms and their ability to form rings, the number of possible cyclic structures, even of small size numbers in the many billions.

The Tiffeneau–Demjanov rearrangement (TDR) is the chemical reaction of a 1-aminomethyl-cycloalkanol with nitrous acid to form an enlarged cycloketone.
The benzilic acid rearrangement is formally the 1,2-rearrangement of 1,2-diketones to form α-hydroxy–carboxylic acids using a base. This reaction receives its name from the reaction of benzil with potassium hydroxide to form benzilic acid. First performed by Justus von Liebig in 1838, it is the first reported example of a rearrangement reaction. It has become a classic reaction in organic synthesis and has been reviewed many times before. It can be viewed as an intramolecular redox reaction, as one carbon center is oxidized while the other is reduced.
The Ramberg–Bäcklund reaction is an organic reaction converting an α-halo sulfone into an alkene in presence of a base with extrusion of sulfur dioxide. The reaction is named after the two Swedish chemists Ludwig Ramberg and Birger Bäcklund. The carbanion formed by deprotonation gives an unstable episulfone that decomposes with elimination of sulfur dioxide. This elimination step is considered to be a concerted cycloelimination.

The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.

In organic chemistry, cyclopropanation refers to any chemical process which generates cyclopropane rings. It is an important process in modern chemistry as many useful compounds bear this motif; for example pyrethroid insecticides and a number of quinolone antibiotics. However, the high ring strain present in cyclopropanes makes them challenging to produce and generally requires the use of highly reactive species, such as carbenes, ylids and carbanions. Many of the reactions proceed in a cheletropic manner.
In organosilicon chemistry, silyl enol ethers are a class of organic compounds that share the common functional group R3Si−O−CR=CR2, composed of an enolate bonded to a silane through its oxygen end and an ethene group as its carbon end. They are important intermediates in organic synthesis.
The semipinacol rearrangement is a rearrangement reaction in organic chemistry involving a heterosubstituted alcohol of the type R1R2(HO)C–C(X)R3R4. The hetero substituent can be a halogen (Cl, Br, I), a tosylate, a mesylate or a thiol group. This reaction proceeds by removal of the leaving group X forming a carbocation as electron deficient center. One of the adjacent alkyl groups then migrates to the positive carbon in a 1,2-shift. Simultaneously with the shift, a pi bond forms from the oxygen to carbon, assisting in driving the migrating group off its position. The result is a ketone or aldehyde. In another definition all semipinacol rearrangements "share a common reactive species in which an electrophilic carbon center, including but not limited to carbocations, is vicinal to an oxygen-containing carbon and can drive the 1,2-migration of a C–C or C–H bond to terminate the process, generating a carbonyl group ".

Cyclopropanone is an organic compound with molecular formula (CH2)2CO consisting of a cyclopropane carbon framework with a ketone functional group. The parent compound is labile, being highly sensitive toward even weak nucleophiles. Surrogates of cyclopropanone include the ketals.
The vinylcyclopropane rearrangement or vinylcyclopropane-cyclopentene rearrangement is a ring expansion reaction, converting a vinyl-substituted cyclopropane ring into a cyclopentene ring.

Basketane is a polycyclic alkane with the chemical formula C10H12. The name is taken from its structural similarity to a basket shape. Basketane was first synthesized in 1966, independently by Masamune and Dauben and Whalen. A patent application published in 1988 used basketane, which is a hydrocarbon, as a source material in doping thin diamond layers because of the molecule's high vapor pressure, carbon ring structure, and fewer hydrogen-to-carbon bond ratio.
The Buchner ring expansion is a two-step organic C-C bond forming reaction used to access 7-membered rings. The first step involves formation of a carbene from ethyl diazoacetate, which cyclopropanates an aromatic ring. The ring expansion occurs in the second step, with an electrocyclic reaction opening the cyclopropane ring to form the 7-membered ring.
Rearrangements, especially those that can participate in cascade reactions, such as the aza-Cope rearrangements, are of high practical as well as conceptual importance in organic chemistry, due to their ability to quickly build structural complexity out of simple starting materials. The aza-Cope rearrangements are examples of heteroatom versions of the Cope rearrangement, which is a [3,3]-sigmatropic rearrangement that shifts single and double bonds between two allylic components. In accordance with the Woodward-Hoffman rules, thermal aza-Cope rearrangements proceed suprafacially. Aza-Cope rearrangements are generally classified by the position of the nitrogen in the molecule :
In organic chemistry, the oxy-Cope rearrangement is a chemical reaction. It involves reorganization of the skeleton of certain unsaturated alcohols. It is a variation of the Cope rearrangement in which 1,5-dien-3-ols are converted to unsaturated carbonyl compounds by a mechanism typical for such a [3,3]-sigmatropic rearrangement.







