Cladistics is an approach to biological classification in which organisms are categorized in groups ("clades") based on hypotheses of most recent common ancestry. The evidence for hypothesized relationships is typically shared derived characteristics (synapomorphies) that are not present in more distant groups and ancestors. However, from an empirical perspective, common ancestors are inferences based on a cladistic hypothesis of relationships of taxa whose character states can be observed. Theoretically, a last common ancestor and all its descendants constitute a (minimal) clade. Importantly, all descendants stay in their overarching ancestral clade. For example, if the terms worms or fishes were used within a strict cladistic framework, these terms would include humans. Many of these terms are normally used paraphyletically, outside of cladistics, e.g. as a 'grade', which are fruitless to precisely delineate, especially when including extinct species. Radiation results in the generation of new subclades by bifurcation, but in practice sexual hybridization may blur very closely related groupings.
In biology, phylogenetics is the study of the evolutionary history and relationships among or within groups of organisms. These relationships are determined by phylogenetic inference, methods that focus on observed heritable traits, such as DNA sequences, protein amino acid sequences, or morphology. The result of such an analysis is a phylogenetic tree—a diagram containing a hypothesis of relationships that reflects the evolutionary history of a group of organisms.
Paraphyly is a taxonomic term describing a grouping that consists of the grouping's last common ancestor and some but not all of its descendant lineages. The grouping is said to be paraphyletic with respect to the excluded subgroups. In contrast, a monophyletic grouping includes a common ancestor and all of its descendants.
A phylogenetic tree, phylogeny or evolutionary tree is a graphical representation which shows the evolutionary history between a set of species or taxa during a specific time. In other words, it is a branching diagram or a tree showing the evolutionary relationships among various biological species or other entities based upon similarities and differences in their physical or genetic characteristics. In evolutionary biology, all life on Earth is theoretically part of a single phylogenetic tree, indicating common ancestry. Phylogenetics is the study of phylogenetic trees. The main challenge is to find a phylogenetic tree representing optimal evolutionary ancestry between a set of species or taxa. Computational phylogenetics focuses on the algorithms involved in finding optimal phylogenetic tree in the phylogenetic landscape.
Molecular phylogenetics is the branch of phylogeny that analyzes genetic, hereditary molecular differences, predominantly in DNA sequences, to gain information on an organism's evolutionary relationships. From these analyses, it is possible to determine the processes by which diversity among species has been achieved. The result of a molecular phylogenetic analysis is expressed in a phylogenetic tree. Molecular phylogenetics is one aspect of molecular systematics, a broader term that also includes the use of molecular data in taxonomy and biogeography.

In cladistics or phylogenetics, an outgroup is a more distantly related group of organisms that serves as a reference group when determining the evolutionary relationships of the ingroup, the set of organisms under study, and is distinct from sociological outgroups. The outgroup is used as a point of comparison for the ingroup and specifically allows for the phylogeny to be rooted. Because the polarity (direction) of character change can be determined only on a rooted phylogeny, the choice of outgroup is essential for understanding the evolution of traits along a phylogeny.
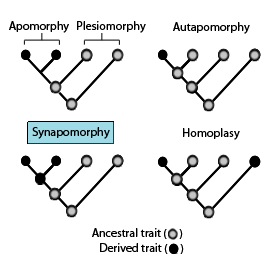
In phylogenetics, an apomorphy is a novel character or character state that has evolved from its ancestral form. A synapomorphy is an apomorphy shared by two or more taxa and is therefore hypothesized to have evolved in their most recent common ancestor. In cladistics, synapomorphy implies homology.
A phylogenetic network is any graph used to visualize evolutionary relationships between nucleotide sequences, genes, chromosomes, genomes, or species. They are employed when reticulation events such as hybridization, horizontal gene transfer, recombination, or gene duplication and loss are believed to be involved. They differ from phylogenetic trees by the explicit modeling of richly linked networks, by means of the addition of hybrid nodes instead of only tree nodes. Phylogenetic trees are a subset of phylogenetic networks. Phylogenetic networks can be inferred and visualised with software such as SplitsTree, the R-package, phangorn, and, more recently, Dendroscope. A standard format for representing phylogenetic networks is a variant of Newick format which is extended to support networks as well as trees.
Computational phylogenetics, phylogeny inference, or phylogenetic inference focuses on computational and optimization algorithms, heuristics, and approaches involved in phylogenetic analyses. The goal is to find a phylogenetic tree representing optimal evolutionary ancestry between a set of genes, species, or taxa. Maximum likelihood, parsimony, Bayesian, and minimum evolution are typical optimality criteria used to assess how well a phylogenetic tree topology describes the sequence data. Nearest Neighbour Interchange (NNI), Subtree Prune and Regraft (SPR), and Tree Bisection and Reconnection (TBR), known as tree rearrangements, are deterministic algorithms to search for optimal or the best phylogenetic tree. The space and the landscape of searching for the optimal phylogenetic tree is known as phylogeny search space.
Perfect phylogeny is a term used in computational phylogenetics to denote a phylogenetic tree in which all internal nodes may be labeled such that all characters evolve down the tree without homoplasy. That is, characteristics do not hold to evolutionary convergence, and do not have analogous structures. Statistically, this can be represented as an ancestor having state "0" in all characteristics where 0 represents a lack of that characteristic. Each of these characteristics changes from 0 to 1 exactly once and never reverts to state 0. It is rare that actual data adheres to the concept of perfect phylogeny.
Bayesian inference of phylogeny combines the information in the prior and in the data likelihood to create the so-called posterior probability of trees, which is the probability that the tree is correct given the data, the prior and the likelihood model. Bayesian inference was introduced into molecular phylogenetics in the 1990s by three independent groups: Bruce Rannala and Ziheng Yang in Berkeley, Bob Mau in Madison, and Shuying Li in University of Iowa, the last two being PhD students at the time. The approach has become very popular since the release of the MrBayes software in 2001, and is now one of the most popular methods in molecular phylogenetics.
Phylogenetic comparative methods (PCMs) use information on the historical relationships of lineages (phylogenies) to test evolutionary hypotheses. The comparative method has a long history in evolutionary biology; indeed, Charles Darwin used differences and similarities between species as a major source of evidence in The Origin of Species. However, the fact that closely related lineages share many traits and trait combinations as a result of the process of descent with modification means that lineages are not independent. This realization inspired the development of explicitly phylogenetic comparative methods. Initially, these methods were primarily developed to control for phylogenetic history when testing for adaptation; however, in recent years the use of the term has broadened to include any use of phylogenies in statistical tests. Although most studies that employ PCMs focus on extant organisms, many methods can also be applied to extinct taxa and can incorporate information from the fossil record.

SplitsTree is a popular freeware program for inferring phylogenetic trees, phylogenetic networks, or, more generally, splits graphs, from various types of data such as a sequence alignment, a distance matrix or a set of trees. SplitsTree implements published methods such as split decomposition, neighbor-net, consensus networks, super networks methods or methods for computing hybridization or simple recombination networks. It uses the NEXUS file format. The splits graph is defined using a special data block.
A supertree is a single phylogenetic tree assembled from a combination of smaller phylogenetic trees, which may have been assembled using different datasets or a different selection of taxa. Supertree algorithms can highlight areas where additional data would most usefully resolve any ambiguities. The input trees of a supertree should behave as samples from the larger tree.
In bioinformatics, alignment-free sequence analysis approaches to molecular sequence and structure data provide alternatives over alignment-based approaches.

Cospeciation is a form of coevolution in which the speciation of one species dictates speciation of another species and is most commonly studied in host-parasite relationships. In the case of a host-parasite relationship, if two hosts of the same species get within close proximity of each other, parasites of the same species from each host are able to move between individuals and mate with the parasites on the other host. However, if a speciation event occurs in the host species, the parasites will no longer be able to "cross over" because the two new host species no longer mate and, if the speciation event is due to a geographic separation, it is very unlikely the two hosts will interact at all with each other. The lack of proximity between the hosts ultimately prevents the populations of parasites from interacting and mating. This can ultimately lead to speciation within the parasite.

The Colubroides are a clade in the suborder Serpentes (snakes). It contains over 85% of all the extant species of snakes. The largest family is Colubridae, but it also includes at least six other families, at least four of which were once classified as "Colubridae" before molecular phylogenetics helped in understanding their relationships. It has been found to be monophyletic.

In phylogenetics, reconciliation is an approach to connect the history of two or more coevolving biological entities. The general idea of reconciliation is that a phylogenetic tree representing the evolution of an entity can be drawn within another phylogenetic tree representing an encompassing entity to reveal their interdependence and the evolutionary events that have marked their shared history. The development of reconciliation approaches started in the 1980s, mainly to depict the coevolution of a gene and a genome, and of a host and a symbiont, which can be mutualist, commensalist or parasitic. It has also been used for example to detect horizontal gene transfer, or understand the dynamics of genome evolution.
