Combinatorial chemistry comprises chemical synthetic methods that make it possible to prepare a large number of compounds in a single process. These compound libraries can be made as mixtures, sets of individual compounds or chemical structures generated by computer software. Combinatorial chemistry can be used for the synthesis of small molecules and for peptides.
A polyamide is a polymer with repeating units linked by amide bonds.

A protecting group or protective group is introduced into a molecule by chemical modification of a functional group to obtain chemoselectivity in a subsequent chemical reaction. It plays an important role in multistep organic synthesis.
Native Chemical Ligation (NCL) is an important extension of the chemical ligation concept for constructing a larger polypeptide chain by the covalent condensation of two or more unprotected peptides segments. Native chemical ligation is the most effective method for synthesizing native or modified proteins of typical size.

Robert Bruce Merrifield was an American biochemist who won the Nobel Prize in Chemistry in 1984 for the invention of solid phase peptide synthesis.

In organic chemistry, peptide synthesis is the production of peptides, compounds where multiple amino acids are linked via amide bonds, also known as peptide bonds. Peptides are chemically synthesized by the condensation reaction of the carboxyl group of one amino acid to the amino group of another. Protecting group strategies are usually necessary to prevent undesirable side reactions with the various amino acid side chains. Chemical peptide synthesis most commonly starts at the carboxyl end of the peptide (C-terminus), and proceeds toward the amino-terminus (N-terminus). Protein biosynthesis in living organisms occurs in the opposite direction.
A lactam is a cyclic amide, formally derived from an amino alkanoic acid. The term is a portmanteau of the words lactone + amide.

Di-tert-butyl dicarbonate is a reagent widely used in organic synthesis. Since this compound can be regarded formally as the acid anhydride derived from a tert-butoxycarbonyl (Boc) group, it is commonly referred to as Boc anhydride. This pyrocarbonate reacts with amines to give N-tert-butoxycarbonyl or so-called Boc derivatives. These carbamate derivatives do not behave as amines, which allows certain subsequent transformations to occur that would be incompatible with the amine functional group. The Boc group can later be removed from the amine using moderately strong acids. Thus, Boc serves as a protective group, for instance in solid phase peptide synthesis. Boc-protected amines are unreactive to most bases and nucleophiles, allowing for the use of the fluorenylmethyloxycarbonyl group (Fmoc) as an orthogonal protecting group.

The tert-butyloxycarbonyl protecting group or tert-butoxycarbonyl protecting group is a protecting group used in organic synthesis.
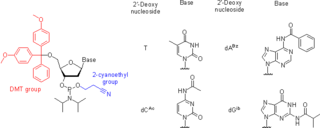
Nucleoside phosphoramidites are derivatives of natural or synthetic nucleosides. They are used to synthesize oligonucleotides, relatively short fragments of nucleic acid and their analogs. Nucleoside phosphoramidites were first introduced in 1981 by Beaucage and Caruthers. To avoid undesired side reactions, reactive hydroxy and exocyclic amino groups present in natural or synthetic nucleosides are appropriately protected. As long as a nucleoside analog contains at least one hydroxy group, the use of the appropriate protecting strategy allows one to convert that to the respective phosphoramidite and to incorporate the latter into synthetic nucleic acids. To be incorporated in the middle of an oligonucleotide chain using phosphoramidite strategy, the nucleoside analog must possess two hydroxy groups or, less often, a hydroxy group and another nucleophilic group (amino or mercapto). Examples include, but are not limited to, alternative nucleotides, LNA, morpholino, nucleosides modified at the 2'-position (OMe, protected NH2, F), nucleosides containing non-canonical bases (hypoxanthine and xanthine contained in natural nucleosides inosine and xanthosine, respectively, tricyclic bases such as G-clamp, etc.) or bases derivatized with a fluorescent group or a linker arm.

Pseudoproline derivatives are artificially created dipeptides to minimize aggregation during Fmoc solid-phase synthesis of peptides.
Oligonucleotide synthesis is the chemical synthesis of relatively short fragments of nucleic acids with defined chemical structure (sequence). The technique is extremely useful in current laboratory practice because it provides a rapid and inexpensive access to custom-made oligonucleotides of the desired sequence. Whereas enzymes synthesize DNA and RNA only in a 5' to 3' direction, chemical oligonucleotide synthesis does not have this limitation, although it is most often carried out in the opposite, 3' to 5' direction. Currently, the process is implemented as solid-phase synthesis using phosphoramidite method and phosphoramidite building blocks derived from protected 2'-deoxynucleosides, ribonucleosides, or chemically modified nucleosides, e.g. LNA or BNA.
Richard A. Houghten is a heterocyclic organic chemist and founder of the journal Peptide Research, which was later merged with the International Journal of Peptide and Protein Research, to become the Journal of Peptide Research. His work mainly concerns peptide activity and pharmacology. He is the founder and president of the Torrey Pines Institute for Molecular Studies (TPIMS), a biomedical research institute. Houghten pioneered the "tea-bag" approach of producing peptides for pharmacological work.
Morten Peter Meldal is a Danish chemist and Nobel laureate. He is a professor of chemistry at the University of Copenhagen in Copenhagen, Denmark. He is best known for developing the CuAAC-click reaction, concurrently with but independent of Valery V. Fokin and K. Barry Sharpless.
Glycopeptides are peptides that contain carbohydrate moieties (glycans) covalently attached to the side chains of the amino acid residues that constitute the peptide.
DNA-encoded chemical libraries (DEL) is a technology for the synthesis and screening on an unprecedented scale of collections of small molecule compounds. DEL is used in medicinal chemistry to bridge the fields of combinatorial chemistry and molecular biology. The aim of DEL technology is to accelerate the drug discovery process and in particular early phase discovery activities such as target validation and hit identification.
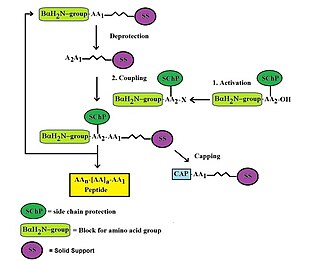
Custom peptide synthesis is the commercial production of peptides for use in biochemistry, biology, biotechnology, pharmacology and molecular medicine. Custom peptide synthesis provides synthetic peptides as valuable tools to biomedical laboratories. Synthetic oligopeptides are used extensively in research for structure-function analysis, for the development of binding assays, the study of receptor agonist/antagonists or as immunogens for the production of specific antibodies. Generally, peptides are synthesized by coupling the carboxyl group or C-terminus of one amino acid to the amino group or N-terminus of another using automated solid phase peptide synthesis chemistries. However, liquid phase synthesis may also be used for specific needs.

The Bailey peptide synthesis is a name reaction in organic chemistry developed 1949 by J. L. Bailey. It is a method for the synthesis of a peptide from α-amino acid-N-carboxylic acid anhydrides (NCAs) and amino acids or peptide esters. The reaction is characterized by short reaction times and a high yield of the target peptide.
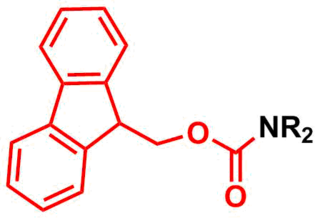
The fluorenylmethoxycarbonyl protecting group (Fmoc) is a base-labile protecting group used in organic synthesis.
The split and pool (split-mix) synthesis is a method in combinatorial chemistry that can be used to prepare combinatorial compound libraries. It is a stepwise, highly efficient process realized in repeated cycles. The procedure makes it possible to prepare millions or even trillions of compounds as mixtures that can be used in drug research.