
An alpha helix is a sequence of amino acids in a protein that are twisted into a coil.
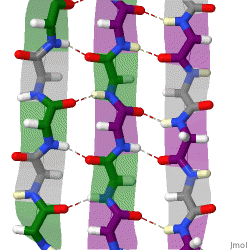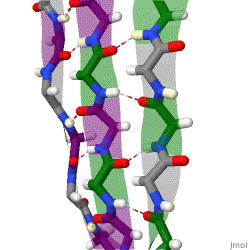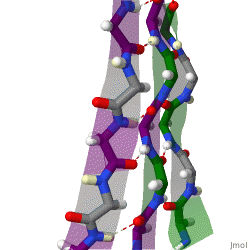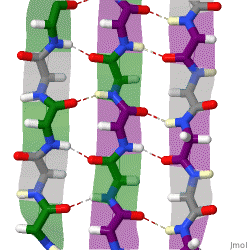
The beta sheet is a common motif of the regular protein secondary structure. Beta sheets consist of beta strands (β-strands) connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A β-strand is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an extended conformation. The supramolecular association of β-sheets has been implicated in the formation of the fibrils and protein aggregates observed in amyloidosis, Alzheimer's disease and other proteinopathies.

In organic chemistry, a peptide bond is an amide type of covalent chemical bond linking two consecutive alpha-amino acids from C1 of one alpha-amino acid and N2 of another, along a peptide or protein chain.

Protein secondary structure is the local spatial conformation of the polypeptide backbone excluding the side chains. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.
Proline (symbol Pro or P) is an organic acid classed as a proteinogenic amino acid (used in the biosynthesis of proteins), although it does not contain the amino group -NH
2 but is rather a secondary amine. The secondary amine nitrogen is in the protonated form (NH2+) under biological conditions, while the carboxyl group is in the deprotonated −COO− form. The "side chain" from the α carbon connects to the nitrogen forming a pyrrolidine loop, classifying it as a aliphatic amino acid. It is non-essential in humans, meaning the body can synthesize it from the non-essential amino acid L-glutamate. It is encoded by all the codons starting with CC (CCU, CCC, CCA, and CCG).

In biochemistry, a Ramachandran plot, originally developed in 1963 by G. N. Ramachandran, C. Ramakrishnan, and V. Sasisekharan, is a way to visualize energetically allowed regions for backbone dihedral angles ψ against φ of amino acid residues in protein structure. The figure on the left illustrates the definition of the φ and ψ backbone dihedral angles. The ω angle at the peptide bond is normally 180°, since the partial-double-bond character keeps the peptide bond planar. The figure in the top right shows the allowed φ,ψ backbone conformational regions from the Ramachandran et al. 1963 and 1968 hard-sphere calculations: full radius in solid outline, reduced radius in dashed, and relaxed tau (N-Cα-C) angle in dotted lines. Because dihedral angle values are circular and 0° is the same as 360°, the edges of the Ramachandran plot "wrap" right-to-left and bottom-to-top. For instance, the small strip of allowed values along the lower-left edge of the plot are a continuation of the large, extended-chain region at upper left.

Protein structure is the three-dimensional arrangement of atoms in an amino acid-chain molecule. Proteins are polymers – specifically polypeptides – formed from sequences of amino acids, which are the monomers of the polymer. A single amino acid monomer may also be called a residue, which indicates a repeating unit of a polymer. Proteins form by amino acids undergoing condensation reactions, in which the amino acids lose one water molecule per reaction in order to attach to one another with a peptide bond. By convention, a chain under 30 amino acids is often identified as a peptide, rather than a protein. To be able to perform their biological function, proteins fold into one or more specific spatial conformations driven by a number of non-covalent interactions, such as hydrogen bonding, ionic interactions, Van der Waals forces, and hydrophobic packing. To understand the functions of proteins at a molecular level, it is often necessary to determine their three-dimensional structure. This is the topic of the scientific field of structural biology, which employs techniques such as X-ray crystallography, NMR spectroscopy, cryo-electron microscopy (cryo-EM) and dual polarisation interferometry, to determine the structure of proteins.
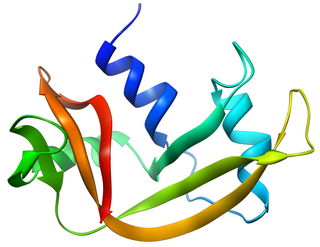
Bovine pancreatic ribonuclease, also often referred to as bovine pancreatic ribonuclease A or simply RNase A, is a pancreatic ribonuclease enzyme that cleaves single-stranded RNA. Bovine pancreatic ribonuclease is one of the classic model systems of protein science. Two Nobel Prizes in Chemistry have been awarded in recognition of work on bovine pancreatic ribonuclease: in 1972, the Prize was awarded to Christian Anfinsen for his work on protein folding and to Stanford Moore and William Stein for their work on the relationship between the protein's structure and its chemical mechanism; in 1984, the Prize was awarded to Robert Bruce Merrifield for development of chemical synthesis of proteins.
A beta bulge can be described as a localized disruption of the regular hydrogen bonding of beta sheet by inserting extra residues into one or both hydrogen bonded β-strands.
A polyproline helix is a type of protein secondary structure which occurs in proteins comprising repeating proline residues. A left-handed polyproline II helix is formed when sequential residues all adopt (φ,ψ) backbone dihedral angles of roughly and have trans isomers of their peptide bonds. This PPII conformation is also common in proteins and polypeptides with other amino acids apart from proline. Similarly, a more compact right-handed polyproline I helix is formed when sequential residues all adopt (φ,ψ) backbone dihedral angles of roughly and have cis isomers of their peptide bonds. Of the twenty common naturally occurring amino acids, only proline is likely to adopt the cis isomer of the peptide bond, specifically the X-Pro peptide bond; steric and electronic factors heavily favor the trans isomer in most other peptide bonds. However, peptide bonds that replace proline with another N-substituted amino acid are also likely to adopt the cis isomer.

A 310 helix is a type of secondary structure found in proteins and polypeptides. Of the numerous protein secondary structures present, the 310-helix is the fourth most common type observed; following α-helices, β-sheets and reverse turns. 310-helices constitute nearly 10–15% of all helices in protein secondary structures, and are typically observed as extensions of α-helices found at either their N- or C- termini. Because of the α-helices tendency to consistently fold and unfold, it has been proposed that the 310-helix serves as an intermediary conformation of sorts, and provides insight into the initiation of α-helix folding.

The beta hairpin is a simple protein structural motif involving two beta strands that look like a hairpin. The motif consists of two strands that are adjacent in primary structure, oriented in an antiparallel direction, and linked by a short loop of two to five amino acids. Beta hairpins can occur in isolation or as part of a series of hydrogen bonded strands that collectively comprise a beta sheet.

Alpha sheet is an atypical secondary structure in proteins, first proposed by Linus Pauling and Robert Corey in 1951. The hydrogen bonding pattern in an alpha sheet is similar to that of a beta sheet, but the orientation of the carbonyl and amino groups in the peptide bond units is distinctive; in a single strand, all the carbonyl groups are oriented in the same direction on one side of the pleat, and all the amino groups are oriented in the same direction on the opposite side of the sheet. Thus the alpha sheet accumulates an inherent separation of electrostatic charge, with one edge of the sheet exposing negatively charged carbonyl groups and the opposite edge exposing positively charged amino groups. Unlike the alpha helix and beta sheet, the alpha sheet configuration does not require all component amino acid residues to lie within a single region of dihedral angles; instead, the alpha sheet contains residues of alternating dihedrals in the traditional right-handed (αR) and left-handed (αL) helical regions of Ramachandran space. Although the alpha sheet is only rarely observed in natural protein structures, it has been speculated to play a role in amyloid disease and it was found to be a stable form for amyloidogenic proteins in molecular dynamics simulations. Alpha sheets have also been observed in X-ray crystallography structures of designed peptides.
Peptide plane flipping is a type of conformational change that can occur in proteins by which the dihedral angles of adjacent amino acids undergo large-scale rotations with little displacement of the side chains. The plane flip is defined as a rotation of the dihedral angles φ,ψ at amino acids i and i+1 such that the resulting angles remain in structurally stable regions of Ramachandran space. The key requirement is that the sum of the ψi angle of residue i and the φi+1 angle of residue i+1 remain roughly constant; in effect, the flip is a crankshaft move about the axis defined by the Cα-C¹ and N-Cα bond vectors of the peptide group, which are roughly parallel. As an example, the type I and type II beta turns differ by a simple flip of the central peptide group of the turn.
β turns are the most common form of turns—a type of non-regular secondary structure in proteins that cause a change in direction of the polypeptide chain. They are very common motifs in proteins and polypeptides. Each consists of four amino acid residues. They can be defined in two ways:
- By the possession of an intra-main-chain hydrogen bond between the CO of residue i and the NH of residue i+3;
- By having a distance of less than 7Å between the Cα atoms of residues i and i+3.
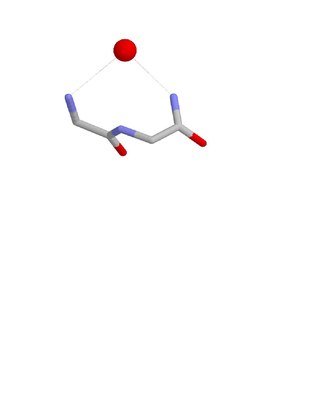
The Nest is a type of protein structural motif. It is a small recurring anion-binding feature of both proteins and peptides. Each consists of the main chain atoms of three consecutive amino acid residues. The main chain NH groups bind the anions while the side chain atoms are often not involved. Proline residues lack NH groups so are rare in nests. About one in 12 of amino acid residues in proteins, on average, belongs to a nest.
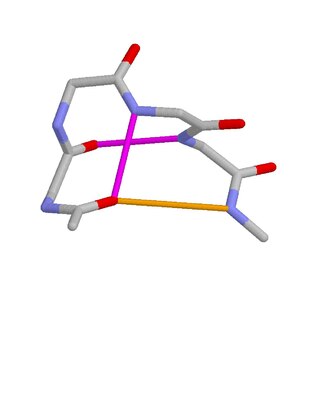
Schellman loops are commonly occurring structural features of proteins and polypeptides. Each has six amino acid residues with two specific inter-mainchain hydrogen bonds and a characteristic main chain dihedral angle conformation. The CO group of residue i is hydrogen-bonded to the NH of residue i+5, and the CO group of residue i+1 is hydrogen-bonded to the NH of residue i+4. Residues i+1, i+2, and i+3 have negative φ (phi) angle values and the phi value of residue i+4 is positive. Schellman loops incorporate a three amino acid residue RL nest, in which three mainchain NH groups form a concavity for hydrogen bonding to carbonyl oxygens. About 2.5% of amino acids in proteins belong to Schellman loops. Two websites are available for examining small motifs in proteins, Motivated Proteins: ; or PDBeMotif:.
The Asx turn is a structural feature in proteins and polypeptides. It consists of three amino acid residues in which residue i is an aspartate (Asp) or asparagine (Asn) that forms a hydrogen bond from its sidechain CO group to the mainchain NH group of residue i+2. About 14% of Asx residues present in proteins belong to Asx turns.
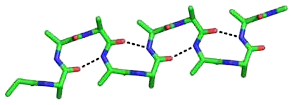
The beta bend ribbon, or beta-bend ribbon, is a structural feature in polypeptides and proteins. The shortest possible has six amino acid residues arranged as two overlapping hydrogen bonded beta turns in which the carbonyl group of residue i is hydrogen-bonded to the NH of residue i+3 while the carbonyl group of residue i+2 is hydrogen-bonded to the NH of residue i+5. In longer ribbons, this bonding is continued in peptides of 8, 10, etc., amino acid residues. A beta bend ribbon can be regarded as an aberrant 310 helix (3/10-helix) that has lost some of its hydrogen bonds. Two websites are available to facilitate finding and examining these features in proteins: Motivated Proteins; and PDBeMotif.
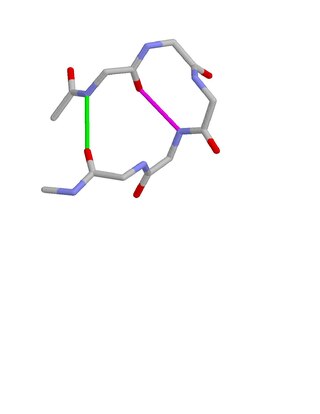
Beta bulge loops are commonly occurring motifs in proteins and polypeptides consisting of five to six amino acids. There are two types: type 1, which is a pentapeptide; and type 2, with six amino acids. They are regarded as a type of beta bulge, and have the alternative name of type G1 beta bulge. Compared to other beta bulges, beta bulge loops give rise to chain reversal such that they often occur at the loop ends of beta hairpins; hairpins of this sort can be described as 3:5 or 4:6. Two websites are available for finding and examining β bulge loops in proteins, Motivated Proteins: and PDBeMotif:.

