
Phenylketonuria (PKU) is an inborn error of metabolism that results in decreased metabolism of the amino acid phenylalanine. Untreated, PKU can lead to intellectual disability, seizures, behavioral problems, and mental disorders. It may also result in a musty smell and lighter skin. A baby born to a mother who has poorly treated PKU may have heart problems, a small head, and low birth weight.
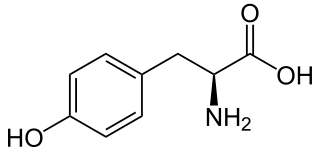
L-Tyrosine or tyrosine or 4-hydroxyphenylalanine is one of the 20 standard amino acids that are used by cells to synthesize proteins. It is a non-essential amino acid with a polar side group. The word "tyrosine" is from the Greek tyrós, meaning cheese, as it was first discovered in 1846 by German chemist Justus von Liebig in the protein casein from cheese. It is called tyrosyl when referred to as a functional group or side chain. While tyrosine is generally classified as a hydrophobic amino acid, it is more hydrophilic than phenylalanine. It is encoded by the codons UAC and UAU in messenger RNA.

Alkaptonuria is a rare inherited genetic disease which is caused by a mutation in the HGD gene for the enzyme homogentisate 1,2-dioxygenase ; if a person inherits an abnormal copy from both parents, the body accumulates an intermediate substance called homogentisic acid in the blood and tissues. Homogentisic acid and its oxidized form alkapton are excreted in the urine, giving it an unusually dark color. The accumulating homogentisic acid causes damage to cartilage and heart valves, as well as precipitating as kidney stones and stones in other organs. Symptoms usually develop in people over 30 years old, although the dark discoloration of the urine is present from birth.

A glycogen storage disease is a metabolic disorder caused by enzyme deficiencies affecting either glycogen synthesis, glycogen breakdown or glycolysis, typically in muscles and/or liver cells.

Phenylalanine hydroxylase (PAH) (EC 1.14.16.1) is an enzyme that catalyzes the hydroxylation of the aromatic side-chain of phenylalanine to generate tyrosine. PAH is one of three members of the biopterin-dependent aromatic amino acid hydroxylases, a class of monooxygenase that uses tetrahydrobiopterin (BH4, a pteridine cofactor) and a non-heme iron for catalysis. During the reaction, molecular oxygen is heterolytically cleaved with sequential incorporation of one oxygen atom into BH4 and phenylalanine substrate. In humans, mutations in its encoding gene, PAH, can lead to the metabolic disorder phenylketonuria.
Inborn errors of metabolism form a large class of genetic diseases involving congenital disorders of enzyme activities. The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). In most of the disorders, problems arise due to accumulation of substances which are toxic or interfere with normal function, or due to the effects of reduced ability to synthesize essential compounds. Inborn errors of metabolism are now often referred to as congenital metabolic diseases or inherited metabolic disorders. To this concept it's possible to include the new term of Enzymopathy. This term was created following the study of Biodynamic Enzymology, a science based on the study of the enzymes and their derivated products. Finally, inborn errors of metabolism were studied for the first time by British physician Archibald Garrod (1857–1936), in 1908. He is known for work that prefigured the "one gene-one enzyme" hypothesis, based on his studies on the nature and inheritance of alkaptonuria. His seminal text, Inborn Errors of Metabolism, was published in 1923.

Homogentisic acid is a phenolic acid usually found in Arbutus unedo (strawberry-tree) honey. It is also present in the bacterial plant pathogen Xanthomonas campestris pv. phaseoli as well as in the yeast Yarrowia lipolytica where it is associated with the production of brown pigments. It is oxidatively dimerised to form hipposudoric acid, one of the main constituents of the 'blood sweat' of hippopotamuses.

4-Hydroxyphenylpyruvate dioxygenase (HPPD), also known as α-ketoisocaproate dioxygenase, is an Fe(II)-containing non-heme oxygenase that catalyzes the second reaction in the catabolism of tyrosine - the conversion of 4-hydroxyphenylpyruvate into homogentisate. HPPD also catalyzes the conversion of phenylpyruvate to 2-hydroxyphenylacetate and the conversion of α-ketoisocaproate to β-hydroxy β-methylbutyrate. HPPD is an enzyme that is found in nearly all aerobic forms of life.

Hawkinsinuria, is an autosomal dominant metabolic disorder affecting the metabolism of tyrosine.

Nitisinone, sold under the brand name Orfadin among others, is a medication used to slow the effects of hereditary tyrosinemia type 1 (HT-1).

ACADSB is a human gene that encodes short/branched chain specific acyl-CoA dehydrogenase (SBCAD), an enzyme in the acyl CoA dehydrogenase family.
Galactose-1-phosphate uridylyltransferase deficiency(classic galactosemia), is the most common type of galactosemia, an inborn error of galactose metabolism, caused by a deficiency of the enzyme galactose-1-phosphate uridylyltransferase. It is an autosomal recessive metabolic disorder that can cause liver disease and death if untreated. Treatment of galactosemia is most successful if initiated early and includes dietary restriction of lactose intake. Because early intervention is key, galactosemia is included in newborn screening programs in many areas. On initial screening, which often involves measuring the concentration of galactose in blood, classic galactosemia may be indistinguishable from other inborn errors of galactose metabolism, including galactokinase deficiency and galactose epimerase deficiency. Further analysis of metabolites and enzyme activities are needed to identify the specific metabolic error.

4-Hydroxyphenylpyruvic acid (4-HPPA) is an intermediate in the metabolism of the amino acid phenylalanine. The aromatic side chain of phenylalanine is hydroxylated by the enzyme phenylalanine hydroxylase to form tyrosine. The conversion from tyrosine to 4-HPPA is in turn catalyzed by tyrosine aminotransferase. Additionally, 4-HPPA can be converted to homogentisic acid which is one of the precursors to ochronotic pigment.

Tyrosine aminotransferase is an enzyme present in the liver and catalyzes the conversion of tyrosine to 4-hydroxyphenylpyruvate.

Fumarylacetoacetase is an enzyme that in humans is encoded by the FAH gene located on chromosome 15. The FAH gene is thought to be involved in the catabolism of the amino acid phenylalanine in humans.

In enzymology, maleylacetoacetate isomerase is an enzyme that catalyzes the chemical reaction

Tyrosinemia type III is a rare disorder caused by a deficiency of the enzyme 4-hydroxyphenylpyruvate dioxygenase, encoded by the gene HPD. This enzyme is abundant in the liver, and smaller amounts are found in the kidneys. It is one of a series of enzymes needed to break down tyrosine. Specifically, 4-hydroxyphenylpyruvate dioxygenase converts a tyrosine byproduct called 4-hydroxyphenylpyruvate to homogentisic acid. Characteristic features of type III tyrosinemia include mild mental retardation, seizures, and periodic loss of balance and coordination. Type III tyrosinemia is very rare; only a few cases have been reported.
Tyrosinemia type II is an autosomal recessive condition with onset between ages 2 and 4 years, when painful circumscribed calluses develop on the pressure points of the palm of the hand and sole of the foot.

Markus Grompe is a professor of Pediatrics and practicing physician at Oregon Health & Science University. since 1991. Since 2004, he has been director of the Oregon Stem Cell Center at OHSU. Until 2018, he was also director of the Papé Family Pediatric Research Institute, Vice Chair for research in the OHSU department of Pediatrics, and holder of the Ray Hickey Endowed Chair at Doernbecher Children's Hospital.

Tyrosinemia type I is a genetic disorder that disrupts the metabolism of the amino acid tyrosine, resulting in damage primarily to the liver along with the kidneys and peripheral nerves. The inability of cells to process tyrosine can lead to chronic liver damage ending in liver failure, as well as renal disease and rickets. Symptoms such as poor growth and enlarged liver are associated with the clinical presentation of the disease. Clinical manifestation of disease occurs typically within the first two years of life. The severity of the disease is correlated with the timing of onset of symptoms, earlier being more severe.


