
Jaundice, also known as icterus, is a yellowish or greenish pigmentation of the skin and sclera due to high bilirubin levels. Jaundice in adults is typically a sign indicating the presence of underlying diseases involving abnormal heme metabolism, liver dysfunction, or biliary-tract obstruction. The prevalence of jaundice in adults is rare, while jaundice in babies is common, with an estimated 80% affected during their first week of life. The most commonly associated symptoms of jaundice are itchiness, pale feces, and dark urine.

In vertebrates, the gallbladder, also known as the cholecyst, is a small hollow organ where bile is stored and concentrated before it is released into the small intestine. In humans, the pear-shaped gallbladder lies beneath the liver, although the structure and position of the gallbladder can vary significantly among animal species. It receives and stores bile, produced by the liver, via the common hepatic duct, and releases it via the common bile duct into the duodenum, where the bile helps in the digestion of fats.

A bile duct is any of a number of long tube-like structures that carry bile, and is present in most vertebrates. The bile duct is separated into three main parts the fundus, body, and neck which are superior, middle, and inferior respectively.

Caroli disease is a rare inherited disorder characterized by cystic dilatation of the bile ducts within the liver. There are two patterns of Caroli disease: focal or simple Caroli disease consists of abnormally widened bile ducts affecting an isolated portion of liver. The second form is more diffuse, and when associated with portal hypertension and congenital hepatic fibrosis, is often referred to as "Caroli syndrome". The underlying differences between the two types are not well understood. Caroli disease is also associated with liver failure and polycystic kidney disease. The disease affects about one in 1,000,000 people, with more reported cases of Caroli syndrome than of Caroli disease.

Autoimmune hepatitis, formerly known as lupoid hepatitis, plasma cell hepatitis, or autoimmune chronic active hepatitis, is a chronic, autoimmune disease of the liver that occurs when the body's immune system attacks liver cells, causing the liver to be inflamed. Common initial symptoms may include fatigue, nausea, muscle aches, or weight loss or signs of acute liver inflammation including fever, jaundice, and right upper quadrant abdominal pain. Individuals with autoimmune hepatitis often have no initial symptoms and the disease may be detected by abnormal liver function tests and increased protein levels during routine bloodwork or the observation of an abnormal-looking liver during abdominal surgery.

Biliary atresia, also known as extrahepatic ductopenia and progressive obliterative cholangiopathy, is a childhood disease of the liver in which one or more bile ducts are abnormally narrow, blocked, or absent. It can be congenital or acquired. It has an incidence of one in 10,000–15,000 live births in the United States, and a prevalence of one in 16,700 in the British Isles. Biliary atresia is most common in East Asia, with a frequency of one in 5,000.

Primary biliary cholangitis (PBC), previously known as primary biliary cirrhosis, is an autoimmune disease of the liver. It results from a slow, progressive destruction of the small bile ducts of the liver, causing bile and other toxins to build up in the liver, a condition called cholestasis. Further slow damage to the liver tissue can lead to scarring, fibrosis, and eventually cirrhosis.

Liver disease, or hepatic disease, is any of many diseases of the liver. If long-lasting it is termed chronic liver disease. Although the diseases differ in detail, liver diseases often have features in common.
Courvoisier's principle states that a painless palpably enlarged gallbladder accompanied with mild jaundice is unlikely to be caused by gallstones. Usually, the term is used to describe the physical examination finding of the right-upper quadrant of the abdomen. This sign implicates possible malignancy of the gallbladder or pancreas and the swelling is unlikely due to gallstones.
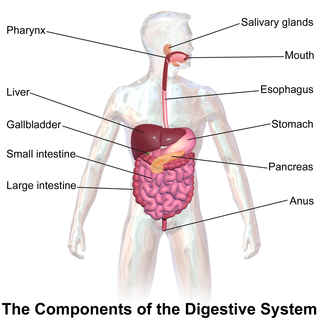
Gastrointestinal diseases refer to diseases involving the gastrointestinal tract, namely the esophagus, stomach, small intestine, large intestine and rectum, and the accessory organs of digestion, the liver, gallbladder, and pancreas.

Alagille syndrome (ALGS) is a genetic disorder that affects primarily the liver and the heart. Problems associated with the disorder generally become evident in infancy or early childhood. The disorder is inherited in an autosomal dominant pattern, and the estimated prevalence of Alagille syndrome is 1 in every 30,000 to 1 in every 40,000 live births. It is named after the French pediatrician Daniel Alagille, who first described the condition in 1969.
Hepatomegaly is enlargement of the liver. It is a non-specific medical sign, having many causes, which can broadly be broken down into infection, hepatic tumours, and metabolic disorder. Often, hepatomegaly presents as an abdominal mass. Depending on the cause, it may sometimes present along with jaundice.
Cholestasis is a condition where the flow of bile from the liver to the duodenum is impaired. The two basic distinctions are:
Cholangiocytes are the epithelial cells of the bile duct. They are cuboidal epithelium in the small interlobular bile ducts, but become columnar and carbonate-secreting in larger bile ducts approaching the porta hepatis and the extrahepatic ducts. They contribute to hepatocyte survival by transporting bile acids.
Progressive familial intrahepatic cholestasis (PFIC) is a group of familial cholestatic conditions caused by defects in biliary epithelial transporters. The clinical presentation usually occurs first in childhood with progressive cholestasis. This usually leads to failure to thrive, cirrhosis, and the need for liver transplantation.
Neonatal cholestasis refers to elevated levels of conjugated bilirubin identified in newborn infants within the first few months of life. Conjugated hyperbilirubinemia is clinically defined as >20% of total serum bilirubin or conjugated bilirubin concentration greater than 1.0 mg/dL regardless of total serum bilirubin concentration. The differential diagnosis for neonatal cholestasis can vary extensively. However, the underlying disease pathology is caused by improper transport and/or defects in excretion of bile from hepatocytes leading to an accumulation of conjugated bilirubin in the body. Generally, symptoms associated with neonatal cholestasis can vary based on the underlying cause of the disease. However, most infants affected will present with jaundice, scleral icterus, failure to thrive, acholic or pale stools, and dark urine.

The biliary tract refers to the liver, gallbladder and bile ducts, and how they work together to make, store and secrete bile. Bile consists of water, electrolytes, bile acids, cholesterol, phospholipids and conjugated bilirubin. Some components are synthesized by hepatocytes ; the rest are extracted from the blood by the liver.

Cirrhosis, also known as liver cirrhosis or hepatic cirrhosis, and end-stage liver disease, is the impaired liver function caused by the formation of scar tissue known as fibrosis due to damage caused by liver disease. Damage to the liver leads to repair of liver tissue and subsequent formation of scar tissue. Over time, scar tissue can replace normal functioning tissue, leading to the impaired liver function of cirrhosis. The disease typically develops slowly over months or years. Early symptoms may include tiredness, weakness, loss of appetite, unexplained weight loss, nausea and vomiting, and discomfort in the right upper quadrant of the abdomen. As the disease worsens, symptoms may include itchiness, swelling in the lower legs, fluid build-up in the abdomen, jaundice, bruising easily, and the development of spider-like blood vessels in the skin. The fluid build-up in the abdomen may develop into spontaneous infections. More serious complications include hepatic encephalopathy, bleeding from dilated veins in the esophagus, stomach, or intestines, and liver cancer.
Hyperbilirubinemia is a clinical condition describing an elevation of blood bilirubin level due to the inability to properly metabolise or excrete bilirubin, a product of erythrocytes breakdown. In severe cases, it is manifested as jaundice, the yellowing of tissues like skin and the sclera when excess bilirubin deposits in them. The US records 52,500 jaundice patients annually. By definition, bilirubin concentration of greater than 3 mg/ml is considered hyperbilirubinemia, following which jaundice progressively develops and becomes apparent when plasma levels reach 20 mg/ml. Rather than a disease itself, hyperbilirubinemia is indicative of multifactorial underlying disorders that trace back to deviations from regular bilirubin metabolism. Diagnosis of hyperbilirubinemia depends on physical examination, urinalysis, serum tests, medical history and imaging to identify the cause. Genetic diseases, alcohol, pregnancy and hepatitis viruses affect the likelihood of hyperbilirubinemia. Causes of hyperbilirubinemia mainly arise from the liver. These include haemolytic anaemias, enzymatic disorders, liver damage and gallstones. Hyperbilirubinemia itself is often benign. Only in extreme cases does kernicterus, a type of brain injury, occur. Therapy for adult hyperbilirubinemia targets the underlying diseases but patients with jaundice often have poor outcomes.
