Related Research Articles

Parkinsonism is a clinical syndrome characterized by tremor, bradykinesia, rigidity, and postural instability. These are the four motor symptoms found in Parkinson's disease (PD) – after which it is named – dementia with Lewy bodies (DLB), Parkinson's disease dementia (PDD), and many other conditions. This set of symptoms occurs in a wide range of conditions and may have many causes, including neurodegenerative conditions, drugs, toxins, metabolic diseases, and neurological conditions other than PD.

Limb–girdle muscular dystrophy (LGMD) is a genetically heterogeneous group of rare muscular dystrophies that share a set of clinical characteristics. It is characterised by progressive muscle wasting which affects predominantly hip and shoulder muscles. LGMD usually has an autosomal pattern of inheritance. It currently has no known cure or treatment.

Frontotemporal dementia (FTD), frontotemporal degeneration disease, or frontotemporal neurocognitive disorder encompasses several types of dementia involving the progressive degeneration of the brain's frontal and temporal lobes. FTDs broadly present as behavioral or language disorders with gradual onsets.
Antisense therapy is a form of treatment that uses antisense oligonucleotides (ASOs) to target messenger RNA (mRNA). ASOs are capable of altering mRNA expression through a variety of mechanisms, including ribonuclease H mediated decay of the pre-mRNA, direct steric blockage, and exon content modulation through splicing site binding on pre-mRNA. Several ASOs have been approved in the United States, the European Union, and elsewhere.
Cerebral atrophy is a common feature of many of the diseases that affect the brain. Atrophy of any tissue means a decrement in the size of the cell, which can be due to progressive loss of cytoplasmic proteins. In brain tissue, atrophy describes a loss of neurons and the connections between them. Brain atrophy can be classified into two main categories: generalized and focal atrophy. Generalized atrophy occurs across the entire brain whereas focal atrophy affects cells in a specific location. If the cerebral hemispheres are affected, conscious thought and voluntary processes may be impaired.
Batten disease is a fatal disease of the nervous system that typically begins in childhood. Onset of symptoms is usually between 5 and 10 years of age. Often, it is autosomal recessive. It is the common name for a group of disorders called the neuronal ceroid lipofuscinoses (NCLs).
Infantile neuronal ceroid lipofuscinoses (INCL) or Santavuori disease or Hagberg-Santavuori disease or Santavuori-Haltia disease or Infantile Finnish type neuronal ceroid lipofuscinosis or Balkan disease is a form of NCL and inherited as a recessive autosomal genetic trait. The disorder is progressive, degenerative and fatal, extremely rare worldwide – with approximately 60 official cases reported by 1982, perhaps 100 with the condition in total today – but relatively common in Finland due to the local founder effect.
Parkinson-plus syndromes (PPS) are a group of neurodegenerative diseases featuring the classical features of Parkinson's disease with additional features that distinguish them from simple idiopathic Parkinson's disease (PD). Parkinson-plus syndromes are either inherited genetically or occur sporadically.
Pantothenate kinase-associated neurodegeneration (PKAN), formerly called Hallervorden–Spatz syndrome, is a genetic degenerative disease of the brain that can lead to parkinsonism, dystonia, dementia, and ultimately death. Neurodegeneration in PKAN is accompanied by an excess of iron that progressively builds up in the brain.

Tauopathies are neurodegenerative diseases involving the aggregation of abnormal tau protein. Tangles are formed by hyperphosphorylation of the microtubule protein known as tau, causing the protein to dissociate from microtubules and form insoluble aggregate. Various neuropathologic phenotypes are identified based on the specific engagement of anatomical regions, cell types, and the presence of unique isoforms of tau within pathological deposits. The designation 'primary tauopathy' is assigned to disorders where the predominant feature is the deposition of tau protein. Alternatively, diseases exhibiting tau pathologies attributed to different and varied underlying causes are termed 'secondary tauopathies. Some neuropathologic phenotypes involving tau protein is Alzheimer's disease, Pick disease, Progressive supranuclear palsy and corticobasal degeneration.
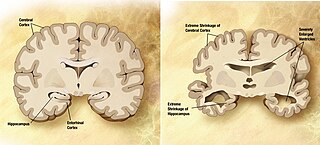
A neurodegenerative disease is caused by the progressive loss of structure or function of neurons, in the process known as neurodegeneration. Such neuronal damage may ultimately involve cell death. Neurodegenerative diseases include amyotrophic lateral sclerosis, multiple sclerosis, Parkinson's disease, Alzheimer's disease, Huntington's disease, multiple system atrophy, tauopathies, and prion diseases. Neurodegeneration can be found in the brain at many different levels of neuronal circuitry, ranging from molecular to systemic. Because there is no known way to reverse the progressive degeneration of neurons, these diseases are considered to be incurable; however research has shown that the two major contributing factors to neurodegeneration are oxidative stress and inflammation. Biomedical research has revealed many similarities between these diseases at the subcellular level, including atypical protein assemblies and induced cell death. These similarities suggest that therapeutic advances against one neurodegenerative disease might ameliorate other diseases as well.
Tourettism refers to the presence of Tourette-like symptoms in the absence of Tourette syndrome, as the result of other diseases or conditions, known as "secondary causes".

Bethlem myopathy is predominantly an autosomal dominant myopathy, classified as a congenital form of limb-girdle muscular dystrophy. There are two types of Bethlem myopathy, based on which type of collagen is affected.

Tripeptidyl-peptidase 1, also known as Lysosomal pepstatin-insensitive protease, is an enzyme that in humans is encoded by the TPP1 gene. TPP1 should not be confused with the TPP1 shelterin protein which protects telomeres and is encoded by the ACD gene. Mutations in the TPP1 gene leads to late infantile neuronal ceroid lipofuscinosis.

Transmembrane protein 106B is a protein that is encoded by the TMEM106B gene. It is found primarily within neurons and oligodendrocytes in the central nervous system with its subcellular location being in lysosomal membranes. TMEM106B helps facilitate important functions for maintaining a healthy lysosome, and therefore certain mutations and polymorphisms can lead to issues with proper lysosomal function. Lysosomes are in charge of clearing out mis-folded proteins and other debris, and thus, play an important role in neurodegenerative diseases that are driven by the accumulation of various mis-folded proteins and aggregates. Due to its impact on lysosomal function, TMEM106B has been investigated and found to be associated to multiple neurodegenerative diseases.

Jansky–Bielschowsky disease is an extremely rare autosomal recessive genetic disorder that is part of the neuronal ceroid lipofuscinosis (NCL) family of neurodegenerative disorders. It is caused by the accumulation of lipopigments in the body due to a deficiency in tripeptidyl peptidase I as a result of a mutation in the TPP1 gene. Symptoms appear between ages 2 and 4 and consist of typical neurodegenerative complications: loss of muscle function (ataxia), drug resistant seizures (epilepsy), apraxia, development of muscle twitches (myoclonus), and vision impairment. This late-infantile form of the disease progresses rapidly once symptoms are onset and ends in death between age 8 and teens. The prevalence of Jansky–Bielschowsky disease is unknown; however, NCL collectively affects an estimated 1 in 100,000 individuals worldwide. Jansky–Bielschowsky disease is related to late-infantile Batten disease and LINCL, and is under the umbrella of neuronal ceroid lipofuscinosis.
Spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME), sometimes called Jankovic–Rivera syndrome, is a very rare neurodegenerative disease whose symptoms include slowly progressive muscle (atrophy), predominantly affecting proximal muscles, combined with denervation and myoclonic seizures. Only 12 known human families are described in scientific literature to have SMA-PME.
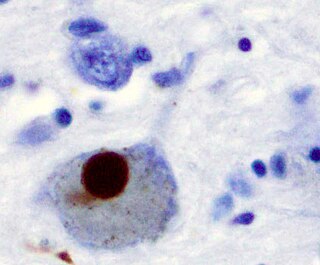
Synucleinopathies are neurodegenerative diseases characterised by the abnormal accumulation of aggregates of alpha-synuclein protein in neurons, nerve fibres or glial cells. There are three main types of synucleinopathy: Parkinson's disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA). Other rare disorders, such as various neuroaxonal dystrophies, also have α-synuclein pathologies. Additionally, autopsy studies have shown that around 6% of sporadic Alzheimer's Disease exhibit α-synuclein positive Lewy pathology, and are sub-classed as Alzheimer's Disease with Amygdalar Restricted Lewy Bodies (AD/ALB).
Muscle–eye–brain (MEB) disease, also known as muscular dystrophy-dystroglycanopathy congenital with brain and eye anomalies A3 (MDDGA3), is a kind of rare congenital muscular dystrophy (CMD), largely characterized by hypotonia at birth. Patients have muscular dystrophy, central nervous system abnormalities and ocular abnormalities. The condition is degenerative.
References
- 1 2 3 4 5 "What is Degenerative Disease". docdoc.com.sg. Archived from the original on 2018-09-17. Retrieved 2018-09-17.
- ↑ "neurodegenerative disorder". National Cancer Institute. 2011-02-02. Archived from the original on 2018-04-23. Retrieved 2018-09-17.
- 1 2 "Neurodegenerative Diseases". Archived from the original on 2018-07-28. Retrieved 2018-09-17.
- 1 2 Nopoulos, PC (2016). "Huntington disease: a single-gene degenerative disorder of the striatum". Dialogues in Clinical Neuroscience. 18 (1): 91–98. doi:10.31887/DCNS.2016.18.1/pnopoulos. ISSN 1294-8322. PMC 4826775 . PMID 27069383.
- 1 2 3 4 Soto, C; Satani, N (2011). "The intricate mechanisms of neurodegeneration in prion diseases". Trends in Molecular Medicine. 17 (1): 14–24. doi:10.1016/j.molmed.2010.09.001. ISSN 1471-4914. PMC 3056171 . PMID 20889378.
- ↑ Patzkó, Á; Shy, ME (2011). "Update on Charcot-Marie-Tooth Disease". Current Neurology and Neuroscience Reports. 11 (1): 78–88. doi:10.1007/s11910-010-0158-7. ISSN 1528-4042. PMC 3685483 . PMID 21080241.
- ↑ Maroon, JC; Winkelman, R; Bost, J; Amos, A; Mathyssek, C; Miele, V (2015-02-11). "Chronic Traumatic Encephalopathy in Contact Sports: A Systematic Review of All Reported Pathological Cases". PLOS ONE. 10 (2): e0117338. Bibcode:2015PLoSO..1017338M. doi: 10.1371/journal.pone.0117338 . ISSN 1932-6203. PMC 4324991 . PMID 25671598.
- ↑ Fraser-Pitt, D; O’Neil, D (2015). "Cystic fibrosis – a multiorgan protein misfolding disease". Future Science OA. 1 (2): FSO57. doi:10.4155/fso.15.57. ISSN 2056-5623. PMC 5137970 . PMID 28031875.
- ↑ "Cytochrome c oxidase deficiency". rarediseases.info.nih.gov. Archived from the original on 2017-10-22. Retrieved 2018-09-17.
- ↑ "Ehlers-Danlos syndromes". rarediseases.info.nih.gov. Archived from the original on 2018-06-16. Retrieved 2018-09-17.
- ↑ Delatycki, M; Williamson, R; Forrest, S (2000). "Friedreich ataxia: an overview". Journal of Medical Genetics. 37 (1): 1–8. doi:10.1136/jmg.37.1.1. ISSN 0022-2593. PMC 1734457 . PMID 10633128.
- ↑ Warren, JD; Rohrer, JD; Rossor, MN (2013-08-06). "Frontotemporal dementia". The BMJ. 347: f4827. doi:10.1136/bmj.f4827. ISSN 0959-8138. PMC 3735339 . PMID 23920254.
- ↑ Barik, R (2016). "Degenerative aortic valve disease and coronary artery disease are either side of a coin". Indian Heart Journal. 68 (3): 432. doi:10.1016/j.ihj.2015.09.010. ISSN 0019-4832. PMC 4911461 . PMID 27316510.
- ↑ Li, H; Zou, Y; Bao, X; Wang, H; Wang, J; Jin, H; Che, Y; Tang, X (2016). "Monozygotic twins with infantile neuroaxonal dystrophy: A case report and literature review". Experimental and Therapeutic Medicine. 12 (5): 3387–3389. doi:10.3892/etm.2016.3761. ISSN 1792-0981. PMC 5103811 . PMID 27882168.
- ↑ Davidson, AE; Hayes, S; Hardcastle, AJ; Tuft, SJ (2014). "The pathogenesis of keratoconus". Eye. 28 (2): 189–195. doi:10.1038/eye.2013.278. ISSN 0950-222X. PMC 3930280 . PMID 24357835.
- ↑ Wallang, BS; Das, S (2013). "Keratoglobus". Eye. 27 (9): 1004–1012. doi:10.1038/eye.2013.130. ISSN 0950-222X. PMC 3772364 . PMID 23807384.
- ↑ van der Knaap, MS; Bugiani, M (2017). "Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms". Acta Neuropathologica. 134 (3): 351–382. doi:10.1007/s00401-017-1739-1. ISSN 0001-6322. PMC 5563342 . PMID 28638987.
- ↑ Anand, A; Sharma, K; Chen, W; Sharma, NK (2014). "Using Current Data to Define New Approach in Age Related Macular Degeneration: Need to Accelerate Translational Research". Current Genomics. 15 (4): 266–277. doi:10.2174/1389202915666140516204512. ISSN 1389-2029. PMC 4133950 . PMID 25132797.
- ↑ Gao, L; Luo, F; Hui, R; Zhou, X (2010). "Recent molecular biological progress in Marfan syndrome and Marfan-associated disorders". Ageing Research Reviews. 9 (3): 363–368. doi:10.1016/j.arr.2009.09.001. ISSN 1568-1637. PMID 19772952. S2CID 205666350.
- ↑ "MELAS Syndrome". NORD (National Organization for Rare Disorders). Archived from the original on 2017-02-20. Retrieved 2018-09-17.
- ↑ El-Hattab, AW; Scaglia, F (2013). "Mitochondrial DNA Depletion Syndromes: Review and Updates of Genetic Basis, Manifestations, and Therapeutic Options". Neurotherapeutics. 10 (2): 186–198. doi:10.1007/s13311-013-0177-6. ISSN 1933-7213. PMC 3625391 . PMID 23385875.
- ↑ Hermena, Shady; Francis, Monica (2021-10-11). "Clinical Presentation, Imaging Features, and Management of Müller–Weiss Disease". Cureus. 13 (10): e18659. doi: 10.7759/cureus.18659 . ISSN 2168-8184. PMC 8579404 . PMID 34786245.
- ↑ Fitzner, D; Simons, M (2010). "Chronic Progressive Multiple Sclerosis – Pathogenesis of Neurodegeneration and Therapeutic Strategies". Current Neuropharmacology. 8 (3): 305–315. doi:10.2174/157015910792246218. ISSN 1570-159X. PMC 3001222 . PMID 21358979.
- ↑ Ubhi, K; Low, P; Masliah, E (2011). "Multiple System Atrophy: A Clinical and Neuropathological Perspective". Trends in Neurosciences. 34 (11): 581–590. doi:10.1016/j.tins.2011.08.003. ISSN 0166-2236. PMC 3200496 . PMID 21962754.
- ↑ Shin, J; Tajrishi, MM; Ogura, Y; Kumar, A (2013). "Wasting Mechanisms in Muscular Dystrophy". The International Journal of Biochemistry & Cell Biology. 45 (10): 2266–2279. doi:10.1016/j.biocel.2013.05.001. ISSN 1357-2725. PMC 3759654 . PMID 23669245.
- ↑ Mole, SE; Williams, Ruth E. (1993). "Neuronal Ceroid-Lipofuscinoses – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY". In Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.). Neuronal Ceroid-Lipofuscinoses. University of Washington, Seattle. PMID 20301601 . Retrieved 2018-09-17.
{{cite book}}:|work=ignored (help) - ↑ Evans, WRH; Hendriksz, CJ (2017). "Niemann–Pick type C disease – the tip of the iceberg? A review of neuropsychiatric presentation, diagnosis and treatment". BJPsych Bulletin. 41 (2): 109–114. doi:10.1192/pb.bp.116.054072. ISSN 2056-4694. PMC 5376728 . PMID 28400970.
- 1 2 Shen, Y; Zhang, Y; Shen, L (2013-04-15). "Postmenopausal women with osteoporosis and osteoarthritis show different microstructural characteristics of trabecular bone in proximal tibia using high-resolution magnetic resonance imaging at 3 tesla". BMC Musculoskeletal Disorders. 14: 136. doi: 10.1186/1471-2474-14-136 . ISSN 1471-2474. PMC 3659090 . PMID 23587336.
- ↑ Bazan, IS; Fares, WH (2015-08-17). "Pulmonary hypertension: diagnostic and therapeutic challenges". Therapeutics and Clinical Risk Management. 11: 1221–1233. doi: 10.2147/TCRM.S74881 . ISSN 1176-6336. PMC 4544628 . PMID 26316767.
- ↑ Owolabi, LF (2013). "Progressive Supranuclear Palsy Misdiagnosed as Parkinson's Disease: A Case Report and Review of Literature". Annals of Medical and Health Sciences Research. 3 (Suppl1): S44–S47. doi: 10.4103/2141-9248.121221 . ISSN 2141-9248. PMC 3853608 . PMID 24349849.
- ↑ Hamel, C (2006-10-11). "Retinitis pigmentosa". Orphanet Journal of Rare Diseases. 1: 40. doi: 10.1186/1750-1172-1-40 . ISSN 1750-1172. PMC 1621055 . PMID 17032466.
- ↑ Abdel-Ahad, P; El Chammai, M; Fneich, A; Issa, R; Kabbara, W; Richa, S (2016). "Les manifestations psychiatriques dans la polyarthrite rhumatoïde". L'Encéphale. 42 (2): 172–176. doi:10.1016/j.encep.2015.12.008. ISSN 0013-7006. PMID 26850214.
- 1 2 Walia, JP; Altaleb, N; Bello, A; Kruck, C; LaFave, MC; Varshney, GK; Burgess, SM; Chowdhury, B; Hurlbut, D (2015). "Long-Term Correction of Sandhoff Disease Following Intravenous Delivery of rAAV9 to Mouse Neonates". Molecular Therapy. 23 (3): 414–422. doi:10.1038/mt.2014.240. ISSN 1525-0016. PMC 4351464 . PMID 25515709.
- ↑ Simone, C; Ramirez, A; Bucchia, M; Rinchetti, P; Rideout, H; Papadimitriou, D; Re, DB; Corti, S (2016). "Is Spinal Muscular Atrophy a disease of the motor neurons only: pathogenesis and therapeutic implications?". Cellular and Molecular Life Sciences. 73 (5): 1003–1020. doi:10.1007/s00018-015-2106-9. ISSN 1420-682X. PMC 4756905 . PMID 26681261.
- ↑ Jafri, SK; Kumar, R; Ibrahim, SH (2018-06-26). "Subacute sclerosing panencephalitis – current perspectives". Pediatric Health, Medicine and Therapeutics. 9: 67–71. doi: 10.2147/PHMT.S126293 . ISSN 1179-9927. PMC 6027681 . PMID 29985487.
- ↑ "Brain Disease Model of Drug & Alcohol Addiction | Hazelden Betty Ford". Archived from the original on 2017-10-27.
- ↑ Korczyn, AD; Vakhapova, V; Grinberg, LT (2012-11-15). "Vascular dementia". Journal of the Neurological Sciences. 322 (1–2): 2–10. doi:10.1016/j.jns.2012.03.027. ISSN 0022-510X. PMC 3435447 . PMID 22575403.
| Authority control databases: National |
|---|