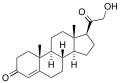
The adrenal cortex is the outer region and also the largest part of an adrenal gland. It is divided into three separate zones: zona glomerulosa, zona fasciculata and zona reticularis. Each zone is responsible for producing specific hormones. It is also a secondary site of androgen synthesis.
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders characterized by impaired cortisol synthesis. It results from the deficiency of one of the five enzymes required for the synthesis of cortisol in the adrenal cortex. Most of these disorders involve excessive or deficient production of hormones such as glucocorticoids, mineralocorticoids, or sex steroids, and can alter development of primary or secondary sex characteristics in some affected infants, children, or adults. It is one of the most common autosomal recessive disorders in humans.
Congenital adrenal hyperplasia due to 17α-hydroxylase deficiency is an uncommon form of congenital adrenal hyperplasia resulting from a defect in the gene CYP17A1, which encodes for the enzyme 17α-hydroxylase. It causes decreased synthesis of cortisol and sex steroids, with resulting increase in mineralocorticoid production. Thus, common symptoms include mild hypocortisolism, ambiguous genitalia in genetic males or failure of the ovaries to function at puberty in genetic females, and hypokalemic hypertension (respectively). However, partial (incomplete) deficiency is notable for having inconsistent symptoms between patients, and affected genetic (XX) females may be wholly asymptomatic except for infertility.
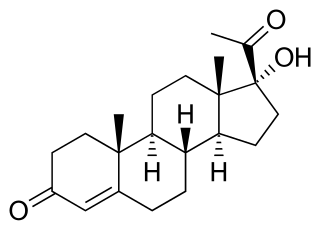
Congenital adrenal hyperplasia due to 21-hydroxylase deficiency, in all its forms, accounts for over 95% of diagnosed cases of congenital adrenal hyperplasia (CAH), and CAH in most contexts refers to 21-hydroxylase deficiency and different mutations related to enzyme impairment have been mapped on protein structure.
The zona glomerulosa of the adrenal gland is the most superficial layer of the adrenal cortex, lying directly beneath the renal capsule. Its cells are ovoid and arranged in clusters or arches.

11-Deoxycorticosterone (DOC), or simply deoxycorticosterone, also known as 21-hydroxyprogesterone, as well as desoxycortone (INN), deoxycortone, and cortexone, is a steroid hormone produced by the adrenal gland that possesses mineralocorticoid activity and acts as a precursor to aldosterone. It is an active (Na+-retaining) mineralocorticoid. As its names indicate, 11-deoxycorticosterone can be understood as the 21-hydroxy-variant of progesterone or as the 11-deoxy-variant of corticosterone.

Cytochrome P450 17A1 is an enzyme of the hydroxylase type that in humans is encoded by the CYP17A1 gene on chromosome 10. It is ubiquitously expressed in many tissues and cell types, including the zona reticularis and zona fasciculata of the adrenal cortex as well as gonadal tissues. It has both 17α-hydroxylase and 17,20-lyase activities, and is a key enzyme in the steroidogenic pathway that produces progestins, mineralocorticoids, glucocorticoids, androgens, and estrogens. More specifically, the enzyme acts upon pregnenolone and progesterone to add a hydroxyl (-OH) group at carbon 17 position (C17) of the steroid D ring, or acts upon 17α-hydroxyprogesterone and 17α-hydroxypregnenolone to split the side-chain off the steroid nucleus.

Cholesterol side-chain cleavage enzyme is commonly referred to as P450scc, where "scc" is an acronym for side-chain cleavage. P450scc is a mitochondrial enzyme that catalyzes conversion of cholesterol to pregnenolone. This is the first reaction in the process of steroidogenesis in all mammalian tissues that specialize in the production of various steroid hormones.

Steroid 21-hydroxylase is an enzyme that hydroxylates steroids at the C21 position and is involved in biosynthesis of aldosterone and cortisol. The enzyme converts progesterone and 17α-hydroxyprogesterone into 11-deoxycorticosterone and 11-deoxycortisol, respectively, within metabolic pathways that ultimately lead to aldosterone and cortisol. Deficiency in the enzyme may cause congenital adrenal hyperplasia.

Steroid 11β-hydroxylase, also known as steroid 11β-monooxygenase, is a steroid hydroxylase found in the zona glomerulosa and zona fasciculata of the adrenal cortex. Named officially the cytochrome P450 11B1, mitochondrial, it is a protein that in humans is encoded by the CYP11B1 gene. The enzyme is involved in the biosynthesis of adrenal corticosteroids by catalyzing the addition of hydroxyl groups during oxidation reactions.
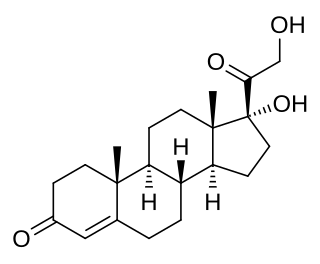
11-Deoxycortisol, also known as cortodoxone (INN), cortexolone as well as 17α,21-dihydroxyprogesterone or 17α,21-dihydroxypregn-4-ene-3,20-dione, is an endogenous glucocorticoid steroid hormone, and a metabolic intermediate towards cortisol. It was first described by Tadeusz Reichstein in 1938 as Substance S, thus has also been referred to as Reichstein's Substance S or Compound S.
Cytochrome P450 reductase is a membrane-bound enzyme required for electron transfer from NADPH to cytochrome P450 and other heme proteins including heme oxygenase in the endoplasmic reticulum of the eukaryotic cell.

18-Hydroxycorticosterone is an endogenous steroid. It is a derivative of corticosterone.
Glucocorticoid remediable aldosteronism also describable as aldosterone synthase hyperactivity, is an autosomal dominant disorder in which the increase in aldosterone secretion produced by ACTH is no longer transient.
A steroidogenesis inhibitor, also known as a steroid biosynthesis inhibitor, is a type of drug which inhibits one or more of the enzymes that are involved in the process of steroidogenesis, the biosynthesis of endogenous steroids and steroid hormones. They may inhibit the production of cholesterol and other sterols, sex steroids such as androgens, estrogens, and progestogens, corticosteroids such as glucocorticoids and mineralocorticoids, and neurosteroids. They are used in the treatment of a variety of medical conditions that depend on endogenous steroids.

21-Deoxycortisol, also known as 11β,17α-dihydroxyprogesterone or as 11β,17α-dihydroxypregn-4-ene-3,20-dione, is a naturally occurring, endogenous steroid related to cortisol (11β,17α,21-trihydroxyprogesterone) which is formed as a metabolite from 17α-hydroxyprogesterone via 11β-hydroxylase.

11β-Hydroxyprogesterone (11β-OHP), also known as 21-deoxycorticosterone, as well as 11β-hydroxypregn-4-ene-3,20-dione, is a naturally occurring, endogenous steroid and derivative of progesterone. It is a potent mineralocorticoid. Syntheses of 11β-OHP from progesterone is catalyzed by the steroid 11β-hydroxylase (CYP11B1) enzyme, and, to a lesser extent, by the aldosterone synthase enzyme (CYP11B2).
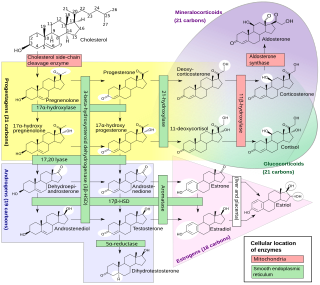
Steroidogenic enzymes are enzymes that are involved in steroidogenesis and steroid biosynthesis. They are responsible for the biosynthesis of the steroid hormones, including sex steroids and corticosteroids, as well as neurosteroids, from cholesterol. Steroidogenic enzymes are most highly expressed in classical steroidogenic tissues, such as the testis, ovary, and adrenal cortex, but are also present in other tissues in the body.

18-Hydroxy-11-deoxycorticosterone is an endogenous steroid and a mineralocorticoid. It is a hydroxylated metabolite of 11-deoxycorticosterone.

The androgen backdoor pathway is a collective name for all metabolic pathways where clinically relevant androgens are synthesized with roundabout of testosterone as an intermediate product. Initially described as pathway where 5α-reduction of 17α-hydroxyprogesterone ultimately leads to 5α-dihydrotestosterone, several other pathways have been since then discovered that lead to 11-oxyandrogens which are potent agonists of the androgen receptors. A backdoor pathway is an alternative to the conventional, canonical androgenic pathway that involves testosterone.