In organic chemistry, an alkene, or olefin, is a hydrocarbon containing a carbon–carbon double bond. The double bond may be internal or in the terminal position. Terminal alkenes are also known as α-olefins.
In chemistry, a nucleophilic substitution is a class of chemical reactions in which an electron-rich chemical species replaces a functional group within another electron-deficient molecule. The molecule that contains the electrophile and the leaving functional group is called the substrate.

An elimination reaction is a type of organic reaction in which two substituents are removed from a molecule in either a one- or two-step mechanism. The one-step mechanism is known as the E2 reaction, and the two-step mechanism is known as the E1 reaction. The numbers refer not to the number of steps in the mechanism, but rather to the kinetics of the reaction: E2 is bimolecular (second-order) while E1 is unimolecular (first-order). In cases where the molecule is able to stabilize an anion but possesses a poor leaving group, a third type of reaction, E1CB, exists. Finally, the pyrolysis of xanthate and acetate esters proceed through an "internal" elimination mechanism, the Ei mechanism.
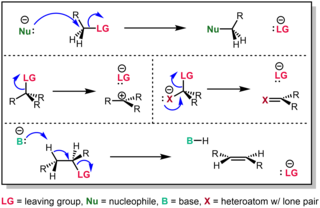
In chemistry, a leaving group is defined by the IUPAC as an atom or group of atoms that detaches from the main or residual part of a substrate during a reaction or elementary step of a reaction. However, in common usage, the term is often limited to a fragment that departs with a pair of electrons in heterolytic bond cleavage. In this usage, a leaving group is a less formal but more commonly used synonym of the term nucleofuge. In this context, leaving groups are generally anions or neutral species, departing from neutral or cationic substrates, respectively, though in rare cases, cations leaving from a dicationic substrate are also known.
Bimolecular nucleophilic substitution (SN2) is a type of reaction mechanism that is common in organic chemistry. In the SN2 reaction, a strong nucleophile forms a new bond to an sp3-hybridised carbon atom via a backside attack, all while the leaving group detaches from the reaction center in a concerted fashion.
An ylide or ylid is a neutral dipolar molecule containing a formally negatively charged atom (usually a carbanion) directly attached to a heteroatom with a formal positive charge (usually nitrogen, phosphorus or sulfur), and in which both atoms have full octets of electrons. The result can be viewed as a structure in which two adjacent atoms are connected by both a covalent and an ionic bond; normally written X+–Y−. Ylides are thus 1,2-dipolar compounds, and a subclass of zwitterions. They appear in organic chemistry as reagents or reactive intermediates.

In organometallic chemistry, organolithium reagents are chemical compounds that contain carbon–lithium (C–Li) bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.
A sigmatropic reaction in organic chemistry is a pericyclic reaction wherein the net result is one σ-bond is changed to another σ-bond in an uncatalyzed intramolecular reaction. The name sigmatropic is the result of a compounding of the long-established sigma designation from single carbon–carbon bonds and the Greek word tropos, meaning turn. In this type of rearrangement reaction, a substituent moves from one part of a π-bonded system to another part in an intramolecular reaction with simultaneous rearrangement of the π system. True sigmatropic reactions are usually uncatalyzed, although Lewis acid catalysis is possible. Sigmatropic reactions often have transition-metal catalysts that form intermediates in analogous reactions. The most well-known of the sigmatropic rearrangements are the [3,3] Cope rearrangement, Claisen rearrangement, Carroll rearrangement, and the Fischer indole synthesis.
Organopalladium chemistry is a branch of organometallic chemistry that deals with organic palladium compounds and their reactions. Palladium is often used as a catalyst in the reduction of alkenes and alkynes with hydrogen. This process involves the formation of a palladium-carbon covalent bond. Palladium is also prominent in carbon-carbon coupling reactions, as demonstrated in tandem reactions.

The Michaelis–Arbuzov reaction is the chemical reaction of a trivalent phosphorus ester with an alkyl halide to form a pentavalent phosphorus species and another alkyl halide. The picture below shows the most common types of substrates undergoing the Arbuzov reaction; phosphite esters (1) react to form phosphonates (2), phosphonites (3) react to form phosphinates (4) and phosphinites (5) react to form phosphine oxides (6).
The Corey–House synthesis (also called the Corey–Posner–Whitesides–House reaction and other permutations) is an organic reaction that involves the reaction of a lithium diorganylcuprate () with an organic halide or pseudohalide () to form a new alkane, as well as an ill-defined organocopper species and lithium (pseudo)halide as byproducts.

The Barbier reaction is an organometallic reaction between an alkyl halide, a carbonyl group and a metal. The reaction can be performed using magnesium, aluminium, zinc, indium, tin, samarium, barium or their salts. The reaction product is a primary, secondary or tertiary alcohol. The reaction is similar to the Grignard reaction but the crucial difference is that the organometallic species in the Barbier reaction is generated in situ, whereas a Grignard reagent is prepared separately before addition of the carbonyl compound. Unlike many Grignard reagents, the organometallic species generated in a Barbier reaction are unstable and thus cannot be stored or sold commercially. Barbier reactions are nucleophilic addition reactions that involve relatively inexpensive, water insensitive metals or metal compounds. For this reason, it is possible in many cases to run the reaction in water, making the procedure part of green chemistry. In contrast, Grignard reagents and organolithium reagents are highly moisture sensitive and must be used under an inert atmosphere without the presence of water. The Barbier reaction is named after Philippe Barbier, who was Victor Grignard's teacher.
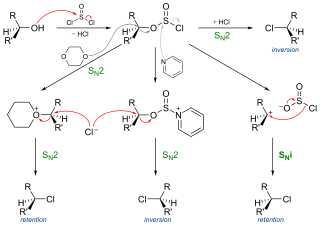
In chemistry, SNi refers to a specific, regio-selective but not often encountered reaction mechanism for nucleophilic aliphatic substitution. The name was introduced by Cowdrey et al. in 1937 to label nucleophilic reactions which occur with retention of configuration, but later was employed to describe various reactions that proceed with a similar mechanism.

The Nicolaou Taxol total synthesis, published by K. C. Nicolaou and his group in 1994 concerns the total synthesis of taxol. Taxol is an important drug in the treatment of cancer but also expensive because the compound is harvested from a scarce resource, namely the pacific yew.

The Johnson–Corey–Chaykovsky reaction is a chemical reaction used in organic chemistry for the synthesis of epoxides, aziridines, and cyclopropanes. It was discovered in 1961 by A. William Johnson and developed significantly by E. J. Corey and Michael Chaykovsky. The reaction involves addition of a sulfur ylide to a ketone, aldehyde, imine, or enone to produce the corresponding 3-membered ring. The reaction is diastereoselective favoring trans substitution in the product regardless of the initial stereochemistry. The synthesis of epoxides via this method serves as an important retrosynthetic alternative to the traditional epoxidation reactions of olefins.
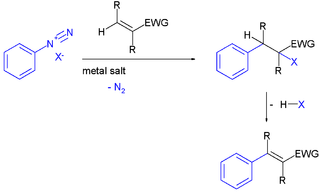
The Meerwein arylation is an organic reaction involving the addition of an aryl diazonium salt (ArN2X) to an electron-poor alkene usually supported by a metal salt. The reaction product is an alkylated arene compound. The reaction is named after Hans Meerwein, one of its inventors who first published it in 1939.

The Mukaiyama taxol total synthesis published by the group of Teruaki Mukaiyama of the Tokyo University of Science between 1997 and 1999 was the 6th successful taxol total synthesis. The total synthesis of Taxol is considered a hallmark in organic synthesis.

The vinyl cation is a carbocation with the positive charge on an alkene carbon. Its empirical formula is C
2H+
3. More generally, a vinylic cation is any disubstituted carbon, where the carbon bearing the positive charge is part of a double bond and is sp hybridized. In the chemical literature, substituted vinylic cations are often referred to as vinyl cations, and understood to refer to the broad class rather than the C
2H+
3 variant alone. The vinyl cation is one of the main types of reactive intermediates involving a non-tetrahedrally coordinated carbon atom, and is necessary to explain a wide variety of observed reactivity trends. Vinyl cations are observed as reactive intermediates in solvolysis reactions, as well during electrophilic addition to alkynes, for example, through protonation of an alkyne by a strong acid. As expected from its sp hybridization, the vinyl cation prefers a linear geometry. Compounds related to the vinyl cation include allylic carbocations and benzylic carbocations, as well as aryl carbocations.
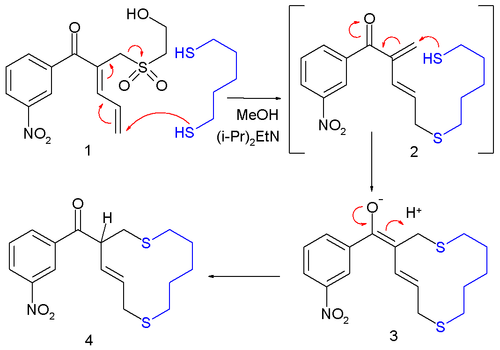
Desulfonylation reactions are chemical reactions leading to the removal of a sulfonyl group from organic compounds. As the sulfonyl functional group is electron-withdrawing, methods for cleaving the sulfur–carbon bonds of sulfones are typically reductive in nature. Olefination or replacement with hydrogen may be accomplished using reductive desulfonylation methods.
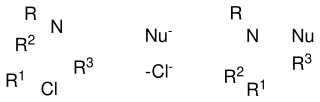
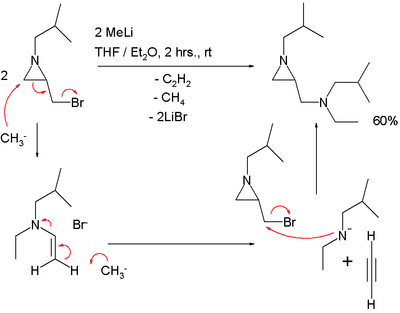
The De Kimpe azirdine synthesis is a name reaction of organic chemistry, for the generation of aziridines by the reaction of α-chloroimines with nucleophiles such as hydride, cyanide, or Grignard reagents.