Chromatin is a complex of DNA and protein found in eukaryotic cells. The primary function is to package long DNA molecules into more compact, denser structures. This prevents the strands from becoming tangled and also plays important roles in reinforcing the DNA during cell division, preventing DNA damage, and regulating gene expression and DNA replication. During mitosis and meiosis, chromatin facilitates proper segregation of the chromosomes in anaphase; the characteristic shapes of chromosomes visible during this stage are the result of DNA being coiled into highly condensed chromatin.
In molecular biology and genetics, transcriptional regulation is the means by which a cell regulates the conversion of DNA to RNA (transcription), thereby orchestrating gene activity. A single gene can be regulated in a range of ways, from altering the number of copies of RNA that are transcribed, to the temporal control of when the gene is transcribed. This control allows the cell or organism to respond to a variety of intra- and extracellular signals and thus mount a response. Some examples of this include producing the mRNA that encode enzymes to adapt to a change in a food source, producing the gene products involved in cell cycle specific activities, and producing the gene products responsible for cellular differentiation in multicellular eukaryotes, as studied in evolutionary developmental biology.
DNA footprinting is a method of investigating the sequence specificity of DNA-binding proteins in vitro. This technique can be used to study protein-DNA interactions both outside and within cells.

Transcriptional repressor CTCF also known as 11-zinc finger protein or CCCTC-binding factor is a transcription factor that in humans is encoded by the CTCF gene. CTCF is involved in many cellular processes, including transcriptional regulation, insulator activity, V(D)J recombination and regulation of chromatin architecture.
ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global binding sites precisely for any protein of interest. Previously, ChIP-on-chip was the most common technique utilized to study these protein–DNA relations.
DNA adenine methyltransferase identification, often abbreviated DamID, is a molecular biology protocol used to map the binding sites of DNA- and chromatin-binding proteins in eukaryotes. DamID identifies binding sites by expressing the proposed DNA-binding protein as a fusion protein with DNA methyltransferase. Binding of the protein of interest to DNA localizes the methyltransferase in the region of the binding site. Adenine methylation does not occur naturally in eukaryotes and therefore adenine methylation in any region can be concluded to have been caused by the fusion protein, implying the region is located near a binding site. DamID is an alternate method to ChIP-on-chip or ChIP-seq.
Paired-end tags (PET) are the short sequences at the 5’ and 3' ends of a DNA fragment which are unique enough that they (theoretically) exist together only once in a genome, therefore making the sequence of the DNA in between them available upon search or upon further sequencing. Paired-end tags (PET) exist in PET libraries with the intervening DNA absent, that is, a PET "represents" a larger fragment of genomic or cDNA by consisting of a short 5' linker sequence, a short 5' sequence tag, a short 3' sequence tag, and a short 3' linker sequence. It was shown conceptually that 13 base pairs are sufficient to map tags uniquely. However, longer sequences are more practical for mapping reads uniquely. The endonucleases used to produce PETs give longer tags but sequences of 50–100 base pairs would be optimal for both mapping and cost efficiency. After extracting the PETs from many DNA fragments, they are linked (concatenated) together for efficient sequencing. On average, 20–30 tags could be sequenced with the Sanger method, which has a longer read length. Since the tag sequences are short, individual PETs are well suited for next-generation sequencing that has short read lengths and higher throughput. The main advantages of PET sequencing are its reduced cost by sequencing only short fragments, detection of structural variants in the genome, and increased specificity when aligning back to the genome compared to single tags, which involves only one end of the DNA fragment.
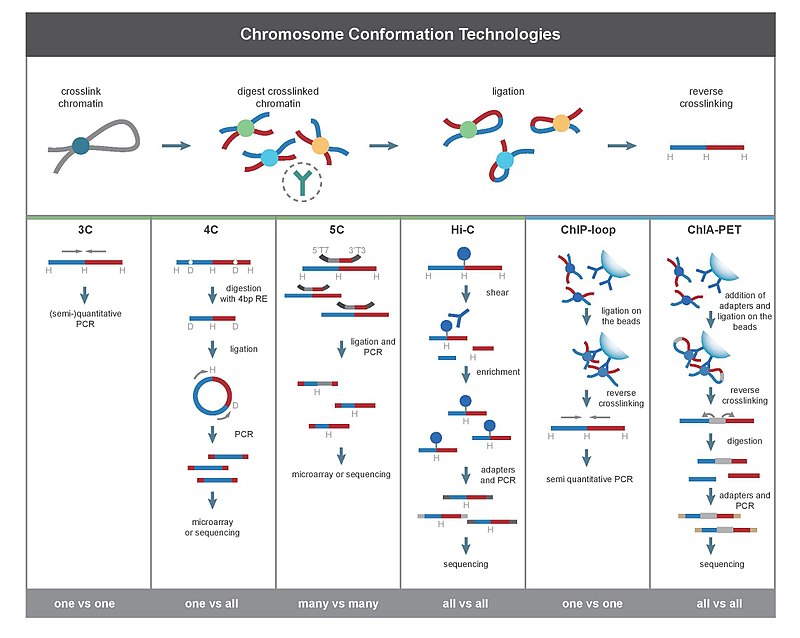
Chromatin Interaction Analysis by Paired-End Tag Sequencing is a technique that incorporates chromatin immunoprecipitation (ChIP)-based enrichment, chromatin proximity ligation, Paired-End Tags, and High-throughput sequencing to determine de novo long-range chromatin interactions genome-wide.

Cas9 is a 160 kilodalton protein which plays a vital role in the immunological defense of certain bacteria against DNA viruses and plasmids, and is heavily utilized in genetic engineering applications. Its main function is to cut DNA and thereby alter a cell's genome. The CRISPR-Cas9 genome editing technique was a significant contributor to the Nobel Prize in Chemistry in 2020 being awarded to Emmanuelle Charpentier and Jennifer Doudna.

STARR-seq is a method to assay enhancer activity for millions of candidates from arbitrary sources of DNA. It is used to identify the sequences that act as transcriptional enhancers in a direct, quantitative, and genome-wide manner.

A topologically associating domain (TAD) is a self-interacting genomic region, meaning that DNA sequences within a TAD physically interact with each other more frequently than with sequences outside the TAD. The median size of a TAD in mouse cells is 880 kb, and they have similar sizes in non-mammalian species. Boundaries at both side of these domains are conserved between different mammalian cell types and even across species and are highly enriched with CCCTC-binding factor (CTCF) and cohesin. In addition, some types of genes appear near TAD boundaries more often than would be expected by chance.
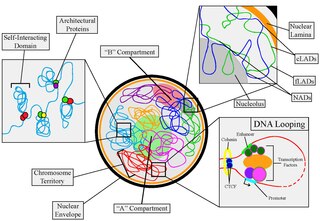
Nuclear organization refers to the spatial distribution of chromatin within a cell nucleus. There are many different levels and scales of nuclear organisation. Chromatin is a higher order structure of DNA.
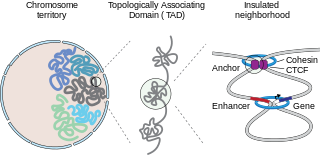
In mammalian biology, insulated neighborhoods are chromosomal loop structures formed by the physical interaction of two DNA loci bound by the transcription factor CTCF and co-occupied by cohesin. Insulated neighborhoods are thought to be structural and functional units of gene control because their integrity is important for normal gene regulation. Current evidence suggests that these structures form the mechanistic underpinnings of higher-order chromosome structures, including topologically associating domains (TADs). Insulated neighborhoods are functionally important in understanding gene regulation in normal cells and dysregulated gene expression in disease.

Single cell epigenomics is the study of epigenomics in individual cells by single cell sequencing. Since 2013, methods have been created including whole-genome single-cell bisulfite sequencing to measure DNA methylation, whole-genome ChIP-sequencing to measure histone modifications, whole-genome ATAC-seq to measure chromatin accessibility and chromosome conformation capture.
In molecular biology, genome architecture mapping (GAM) is a cryosectioning method to map colocalized DNA regions in a ligation independent manner. It overcomes some limitations of Chromosome conformation capture (3C), as these methods have a reliance on digestion and ligation to capture interacting DNA segments. GAM is the first genome-wide method for capturing three-dimensional proximities between any number of genomic loci without ligation.
DXZ4 is a variable number tandemly repeated DNA sequence. In humans it is composed of 3kb monomers containing a highly conserved CTCF binding site. CTCF is a transcription factor protein and the main insulator responsible for partitioning of chromatin domains in the vertebrate genome.
Human epigenome is the complete set of structural modifications of chromatin and chemical modifications of histones and nucleotides. These modifications affect according to cellular type and development status. Various studies show that epigenome depends on exogenous factors.

Hi-C is a high-throughput genomic and epigenomic technique first described in 2009 by Lieberman-Aiden et al. to capture chromatin conformation. In general, Hi-C is considered as a derivative of a series of chromosome conformation capture technologies, including but not limited to 3C, 4C, and 5C. Hi-C comprehensively detects genome-wide chromatin interactions in the cell nucleus by combining 3C and next-generation sequencing (NGS) approaches and has been considered as a qualitative leap in C-technology development and the beginning of 3D genomics.

Proximity ligation-assisted chromatin immunoprecipitation sequencing (PLAC-seq) is a chromatin conformation capture(3C)-based technique to detect and quantify genomic chromatin structure from a protein-centric approach. PLAC-seq combines in situ Hi-C and chromatin immunoprecipitation (ChIP), which allows for the identification of long-range chromatin interactions at a high resolution with low sequencing costs. Mapping long-range 3-dimensional(3D) chromatin interactions is important in identifying transcription enhancers and non-coding variants that can be linked to human diseases.
Pore-C is an emerging genomic technique which utilizes chromatin conformation capture (3C) and Oxford Nanopore Technologies' (ONT) long-read sequencing to characterize three-dimensional (3D) chromatin structure. To characterize concatemers, the originators of Pore-C developed an algorithm to identify alignments that are assigned to a restriction fragment; concatemers with greater than two associated fragments are deemed high order. Pore-C attempts to improve on previous 3C technologies, such as Hi-C and SPRITE, by not requiring DNA amplification prior to sequencing. This technology was developed as a simpler and more easily scalable method of capturing higher-order chromatin structure and mapping regions of chromatin contact. In addition, Pore-C can be used to visualize epigenomic interactions due to the capability of ONT long-read sequencing to detect DNA methylation. Applications of this technology include analysis of combinatorial chromatin interactions, the generation of de novo chromosome scale assemblies, visualization of regions associated with multi-locus histone bodies, and detection and resolution of structural variants.