
An alpha helix is a sequence of amino acids in a protein that are twisted into a coil.

Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.

In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences such as calculating the distance cost between strings in a natural language, or to display financial data.
Grammar theory to model symbol strings originated from work in computational linguistics aiming to understand the structure of natural languages. Probabilistic context free grammars (PCFGs) have been applied in probabilistic modeling of RNA structures almost 40 years after they were introduced in computational linguistics.

Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; it is important in medicine and biotechnology.
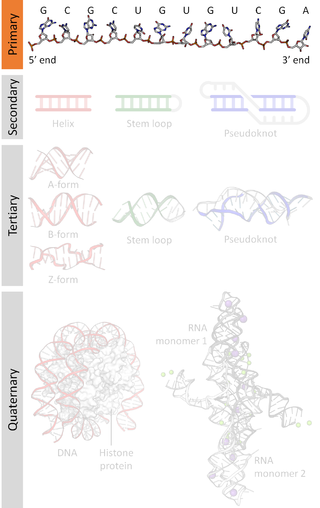
A nucleic acid sequence is a succession of bases within the nucleotides forming alleles within a DNA or RNA (GACU) molecule. This succession is denoted by a series of a set of five different letters that indicate the order of the nucleotides. By convention, sequences are usually presented from the 5' end to the 3' end. For DNA, with its double helix, there are two possible directions for the notated sequence; of these two, the sense strand is used. Because nucleic acids are normally linear (unbranched) polymers, specifying the sequence is equivalent to defining the covalent structure of the entire molecule. For this reason, the nucleic acid sequence is also termed the primary structure.
In bioinformatics, BLAST is an algorithm and program for comparing primary biological sequence information, such as the amino-acid sequences of proteins or the nucleotides of DNA and/or RNA sequences. A BLAST search enables a researcher to compare a subject protein or nucleotide sequence with a library or database of sequences, and identify database sequences that resemble the query sequence above a certain threshold. For example, following the discovery of a previously unknown gene in the mouse, a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene; BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence.
In a chain-like biological molecule, such as a protein or nucleic acid, a structural motif is a common three-dimensional structure that appears in a variety of different, evolutionarily unrelated molecules. A structural motif does not have to be associated with a sequence motif; it can be represented by different and completely unrelated sequences in different proteins or RNA.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
In molecular biology and bioinformatics, the consensus sequence is the calculated sequence of most frequent residues, either nucleotide or amino acid, found at each position in a sequence alignment. It represents the results of multiple sequence alignments in which related sequences are compared to each other and similar sequence motifs are calculated. Such information is important when considering sequence-dependent enzymes such as RNA polymerase.
Computational genomics refers to the use of computational and statistical analysis to decipher biology from genome sequences and related data, including both DNA and RNA sequence as well as other "post-genomic" data. These, in combination with computational and statistical approaches to understanding the function of the genes and statistical association analysis, this field is also often referred to as Computational and Statistical Genetics/genomics. As such, computational genomics may be regarded as a subset of bioinformatics and computational biology, but with a focus on using whole genomes to understand the principles of how the DNA of a species controls its biology at the molecular level and beyond. With the current abundance of massive biological datasets, computational studies have become one of the most important means to biological discovery.
In molecular biology, protein threading, also known as fold recognition, is a method of protein modeling which is used to model those proteins which have the same fold as proteins of known structures, but do not have homologous proteins with known structure. It differs from the homology modeling method of structure prediction as it is used for proteins which do not have their homologous protein structures deposited in the Protein Data Bank (PDB), whereas homology modeling is used for those proteins which do. Threading works by using statistical knowledge of the relationship between the structures deposited in the PDB and the sequence of the protein which one wishes to model.
A position weight matrix (PWM), also known as a position-specific weight matrix (PSWM) or position-specific scoring matrix (PSSM), is a commonly used representation of motifs (patterns) in biological sequences.

Multiple sequence alignment (MSA) is the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. These alignments are used to infer evolutionary relationships via phylogenetic analysis and can highlight homologous features between sequences. Alignments highlight mutation events such as point mutations, insertion mutations and deletion mutations, and alignments are used to assess sequence conservation and infer the presence and activity of protein domains, tertiary structures, secondary structures, and individual amino acids or nucleotides.

Biomolecular structure is the intricate folded, three-dimensional shape that is formed by a molecule of protein, DNA, or RNA, and that is important to its function. The structure of these molecules may be considered at any of several length scales ranging from the level of individual atoms to the relationships among entire protein subunits. This useful distinction among scales is often expressed as a decomposition of molecular structure into four levels: primary, secondary, tertiary, and quaternary. The scaffold for this multiscale organization of the molecule arises at the secondary level, where the fundamental structural elements are the molecule's various hydrogen bonds. This leads to several recognizable domains of protein structure and nucleic acid structure, including such secondary-structure features as alpha helixes and beta sheets for proteins, and hairpin loops, bulges, and internal loops for nucleic acids. The terms primary, secondary, tertiary, and quaternary structure were introduced by Kaj Ulrik Linderstrøm-Lang in his 1951 Lane Medical Lectures at Stanford University.

HMMER is a free and commonly used software package for sequence analysis written by Sean Eddy. Its general usage is to identify homologous protein or nucleotide sequences, and to perform sequence alignments. It detects homology by comparing a profile-HMM to either a single sequence or a database of sequences. Sequences that score significantly better to the profile-HMM compared to a null model are considered to be homologous to the sequences that were used to construct the profile-HMM. Profile-HMMs are constructed from a multiple sequence alignment in the HMMER package using the hmmbuild program. The profile-HMM implementation used in the HMMER software was based on the work of Krogh and colleagues. HMMER is a console utility ported to every major operating system, including different versions of Linux, Windows, and macOS.

Nucleic acid secondary structure is the basepairing interactions within a single nucleic acid polymer or between two polymers. It can be represented as a list of bases which are paired in a nucleic acid molecule. The secondary structures of biological DNAs and RNAs tend to be different: biological DNA mostly exists as fully base paired double helices, while biological RNA is single stranded and often forms complex and intricate base-pairing interactions due to its increased ability to form hydrogen bonds stemming from the extra hydroxyl group in the ribose sugar.

Gary Stormo is an American geneticist and currently Joseph Erlanger Professor in the Department of Genetics and the Center for Genome Sciences and Systems Biology at Washington University School of Medicine in St Louis. He is considered one of the pioneers of bioinformatics and genomics. His research combines experimental and computational approaches in order to identify and predict regulatory sequences in DNA and RNA, and their contributions to the regulatory networks that control gene expression.
PSI-blast based secondary structure PREDiction (PSIPRED) is a method used to investigate protein structure. It uses artificial neural network machine learning methods in its algorithm. It is a server-side program, featuring a website serving as a front-end interface, which can predict a protein's secondary structure from the primary sequence.

An array of protein tandem repeats is defined as several adjacent copies having the same or similar sequence motifs. These periodic sequences are generated by internal duplications in both coding and non-coding genomic sequences. Repetitive units of protein tandem repeats are considerably diverse, ranging from the repetition of a single amino acid to domains of 100 or more residues.

