The complement system, also known as complement cascade, is a part of the immune system that enhances (complements) the ability of antibodies and phagocytic cells to clear microbes and damaged cells from an organism, promote inflammation, and attack the pathogen's cell membrane. It is part of the innate immune system, which is not adaptable and does not change during an individual's lifetime. The complement system can, however, be recruited and brought into action by antibodies generated by the adaptive immune system.
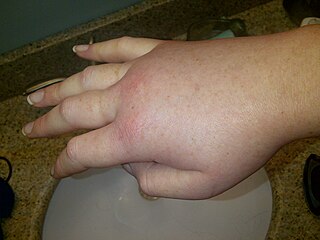
Hereditary angioedema (HAE) is a disorder that results in recurrent attacks of severe swelling. The swelling most commonly affects the arms, legs, face, intestinal tract, and airway. If the intestinal tract is affected, abdominal pain and vomiting may occur. Swelling of the airway can result in its obstruction and trouble breathing. Without preventive treatment, attacks typically occur every two weeks and last for a few days.

Angioedema is an area of swelling (edema) of the lower layer of skin and tissue just under the skin or mucous membranes. The swelling may occur in the face, tongue, larynx, abdomen, or arms and legs. Often it is associated with hives, which are swelling within the upper skin. Onset is typically over minutes to hours.

The classical complement pathway is one of three pathways which activate the complement system, which is part of the immune system. The classical complement pathway is initiated by antigen-antibody complexes with the antibody isotypes IgG and IgM.
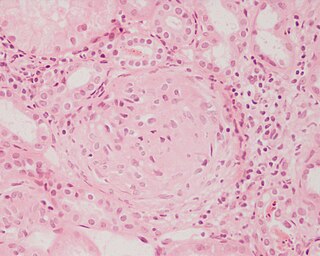
Glomerulonephritis (GN) is a term used to refer to several kidney diseases. Many of the diseases are characterised by inflammation either of the glomeruli or of the small blood vessels in the kidneys, hence the name, but not all diseases necessarily have an inflammatory component.

C1-inhibitor is a protease inhibitor belonging to the serpin superfamily. Its main function is the inhibition of the complement system to prevent spontaneous activation but also as the major regulator of the contact system. C1-inhibitor is an acute-phase protein that circulates in blood at levels of around 0.25 g/L. The levels rise ~2-fold during inflammation. C1-inhibitor irreversibly binds to and inactivates C1r and C1s proteases in the C1 complex of classical pathway of complement. MASP-1 and MASP-2 proteases in MBL complexes of the lectin pathway are also inactivated. This way, C1-inhibitor prevents the proteolytic cleavage of later complement components C4 and C2 by C1 and MBL. Although named after its complement inhibitory activity, C1-inhibitor also inhibits proteases of the fibrinolytic, clotting, and kinin pathways. Note that C1-inhibitor is the most important physiological inhibitor of plasma kallikrein, FXIa, and FXIIa.

Complement component 3, often simply called C3, is a protein of the immune system that is found primarily in the blood. It plays a central role in the complement system of vertebrate animals and contributes to innate immunity. In humans it is encoded on chromosome 19 by a gene called C3.

The lectin pathway or MBL pathway is a type of cascade reaction in the complement system, similar in structure to the classical complement pathway, in that, after activation, it proceeds through the action of C4 and C2 to produce activated complement proteins further down the cascade. In contrast to the classical complement pathway, the lectin pathway does not recognize an antibody bound to its target. The lectin pathway starts with mannose-binding lectin (MBL) or ficolin binding to certain sugars.

Acute proliferative glomerulonephritis is a disorder of the small blood vessels of the kidney. It is a common complication of bacterial infections, typically skin infection by Streptococcus bacteria types 12, 4 and 1 (impetigo) but also after streptococcal pharyngitis, for which it is also known as postinfectious glomerulonephritis (PIGN) or poststreptococcal glomerulonephritis (PSGN). It can be a risk factor for future albuminuria. In adults, the signs and symptoms of infection may still be present at the time when the kidney problems develop, and the terms infection-related glomerulonephritis or bacterial infection-related glomerulonephritis are also used. Acute glomerulonephritis resulted in 19,000 deaths in 2013, down from 24,000 deaths in 1990 worldwide.

Complement C2 is a protein that in humans is encoded by the C2 gene. The protein encoded by this gene is part of the classical pathway of the complement system, acting as a multi-domain serine protease. Deficiency of C2 has been associated with certain autoimmune diseases.

Properdin deficiency is a rare X-linked disease in which properdin, an important complement factor responsible for the stabilization of the alternative C3 convertase, is deficient. There are three forms of properdin deficiencies: Type I, which is identified by the total absence of the properdin protein in the plasma, Type II, which is a low but detectable amount of the properdin protein in the plasma, and Type III, which is a rare case of normal levels of properdin protein, but a dysfunctional variant. One of the first studied cases of properdin deficiency was in 1980 by Davis and Forrestal. These families had members with only partial deficiencies which resulted in a lowered consumption of the C3 protein. Properdin deficiency was studied again shortly after in 1982 by Sjoholm in which all of the subjects were deceased shortly after the study because of their disease. The largest study of properdin deficiency was in 1989 by Fijen which included nine males across three generations. Out of the 46 family members in Fijen's study, the 9 who were affected were found to be more susceptible to diseases from the Neisseria genus.
C3b is the larger of two elements formed by the cleavage of complement component 3, and is considered an important part of the innate immune system. C3b is potent in opsonization: tagging pathogens, immune complexes (antigen-antibody), and apoptotic cells for phagocytosis. Additionally, C3b plays a role in forming a C3 convertase when bound to Factor B, or a C5 convertase when bound to C4b and C2b or when an additional C3b molecule binds to the C3bBb complex.
Barraquer–Simons syndrome is a rare form of lipodystrophy, which usually first affects the head, and then spreads to the thorax. It is named for Luis Barraquer Roviralta (1855–1928), a Spanish physician, and Arthur Simons (1879–1942), a German physician. Some evidence links it to LMNB2.
Complement 2 deficiency is a type of complement deficiency caused by any one of several different alterations in the structure of complement component 2.

Lupus, technically known as systemic lupus erythematosus (SLE), is an autoimmune disease in which the body's immune system mistakenly attacks healthy tissue in many parts of the body. Symptoms vary among people and may be mild to severe. Common symptoms include painful and swollen joints, fever, chest pain, hair loss, mouth ulcers, swollen lymph nodes, feeling tired, and a red rash which is most commonly on the face. Often there are periods of illness, called flares, and periods of remission during which there are few symptoms.

Acquired C1 esterase inhibitor deficiency, also referred to as acquired angioedema (AAE), is a rare medical condition that presents as body swelling that can be life-threatening and manifests due to another underlying medical condition. The acquired form of this disease can occur from a deficiency or abnormal function of the enzyme C1 esterase inhibitor (C1-INH). This disease is also abbreviated in medical literature as C1INH-AAE. This form of angioedema is considered acquired due to its association with lymphatic malignancies, immune system disorders, or infections. Typically, acquired angioedema presents later in adulthood, in contrast to hereditary angioedema which usually presents from early childhood and with similar symptoms.
Diffuse proliferative glomerulonephritis (DPGN) is a type of glomerulonephritis that is the most serious form of renal lesions in SLE and is also the most common, occurring in 35% to 60% of patients. In absence of SLE, DPGN pathology looks more like Membranoproliferative glomerulonephritis
Complement 3 deficiency is a genetic condition affecting complement component 3 (C3). People can suffer from either primary or secondary C3 deficiency. Primary C3 deficiency refers to an inherited autosomal-recessive disorder that involves mutations in the gene for C3. Secondary C3 deficiency results from a lack of factor I or factor H, two proteins that are key for the regulation of C3. Both primary and secondary C3 deficiency are characterized by low levels or absence of C3.
Total complement activity (TCA) refers to a series of tests that determine the functioning of the complement system in an individual.