
Hemolysis or haemolysis, also known by several other names, is the rupturing (lysis) of red blood cells (erythrocytes) and the release of their contents (cytoplasm) into surrounding fluid. Hemolysis may occur in vivo or in vitro.
Aplastic anemia (AA) is a severe hematologic condition in which the body fails to make blood cells in sufficient numbers. Aplastic anemia is associated with cancer and various cancer syndromes. Blood cells are produced in the bone marrow by stem cells that reside there. Aplastic anemia causes a deficiency of all blood cell types: red blood cells, white blood cells, and platelets.
Glycosylphosphatidylinositol or glycophosphatidylinositol (GPI) is a phosphoglyceride that can be attached to the C-terminus of a protein during posttranslational modification. The resulting GPI-anchored proteins play key roles in a wide variety of biological processes. GPI is composed of a phosphatidylinositol group linked through a carbohydrate-containing linker and via an ethanolamine phosphate (EtNP) bridge to the C-terminal amino acid of a mature protein. The two fatty acids within the hydrophobic phosphatidyl-inositol group anchor the protein to the cell membrane.

Hereditary spherocytosis (HS) is a congenital hemolytic disorder wherein a genetic mutation coding for a structural membrane protein phenotype causes the red blood cells to be sphere-shaped (spherocytosis), rather than the normal biconcave disk shape. This abnormal shape interferes with the cells' ability to flex during blood circulation, and also makes them more prone to rupture under osmotic stress, mechanical stress, or both. Cells with the dysfunctional proteins are degraded in the spleen, which leads to a shortage of erythrocytes and results in hemolytic anemia.

Hemolytic anemia or haemolytic anaemia is a form of anemia due to hemolysis, the abnormal breakdown of red blood cells (RBCs), either in the blood vessels or elsewhere in the human body (extravascular). This most commonly occurs within the spleen, but also can occur in the reticuloendothelial system or mechanically. Hemolytic anemia accounts for 5% of all existing anemias. It has numerous possible consequences, ranging from general symptoms to life-threatening systemic effects. The general classification of hemolytic anemia is either intrinsic or extrinsic. Treatment depends on the type and cause of the hemolytic anemia.
Autoimmune hemolytic anemia (AIHA) occurs when antibodies directed against the person's own red blood cells (RBCs) cause them to burst (lyse), leading to an insufficient number of oxygen-carrying red blood cells in the circulation. The lifetime of the RBCs is reduced from the normal 100–120 days to just a few days in serious cases. The intracellular components of the RBCs are released into the circulating blood and into tissues, leading to some of the characteristic symptoms of this condition. The antibodies are usually directed against high-incidence antigens, therefore they also commonly act on allogenic RBCs. AIHA is a relatively rare condition, with an incidence of 5–10 cases per 1 million persons per year in the warm-antibody type and 0.45 to 1.9 cases per 1 million persons per year in the cold antibody type. Autoimmune hemolysis might be a precursor of later onset systemic lupus erythematosus.
Paroxysmal cold hemoglobinuria (PCH) or Donath–Landsteiner hemolytic anemia (DLHA) is an autoimmune hemolytic anemia featured by complement-mediated intravascular hemolysis after cold exposure. It can present as an acute non-recurrent postinfectious event in children, or chronic relapsing episodes in adults with hematological malignancies or tertiary syphilis. Described by Julius Donath (1870–1950) and Karl Landsteiner (1868–1943) in 1904, PCH is one of the first clinical entities recognized as an autoimmune disorder.

Complement decay-accelerating factor, also known as CD55 or DAF, is a protein that, in humans, is encoded by the CD55 gene.
Eculizumab, sold under the brand name Soliris among others, is a recombinant humanized monoclonal antibody used to treat paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), generalized myasthenia gravis, and neuromyelitis optica. In people with PNH, it reduces both the destruction of red blood cells and need for blood transfusion, but does not appear to affect the risk of death. Eculizumab was the first drug approved for each of its uses, and its approval was granted based on small trials. It is given by intravenous infusion.
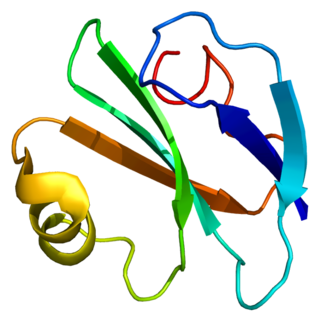
CD59 glycoprotein, also known as MAC-inhibitory protein (MAC-IP), membrane inhibitor of reactive lysis (MIRL), or protectin, is a protein that in humans is encoded by the CD59 gene. It is an LU domain and belongs to the LY6/uPAR/alpha-neurotoxin protein family.

Phosphatidylinositol N-acetylglucosaminyltransferase subunit A is the catalytic subunit of the phosphatidylinositol N-acetylglucosaminyltransferase enzyme, which in humans is encoded by the PIGA gene.
Fluorescein-labeled proaerolysin (FLAER) is used in a flow cytometric assay to diagnose paroxysmal nocturnal hemoglobinuria (PNH). The assay takes advantage of the action of proaerolysin, a prototoxin of aerolysin, a virulence factor of the bacterium Aeromonas hydrophila. Proaerolysin binds to the glycophosphatidylinositol(GPI) anchor in the plasma membrane of cells. Cells affected by PNH lack GPI anchoring proteins, and thus are not bound by proaerolysin. Of note, the FLAER-based assay is not suitable for evaluation of erythrocytes and platelets in PNH but flow cytometry assays based on CD55, CD59 and others are suitable.
The Ham test is a blood test used in the diagnosis of paroxysmal nocturnal hemoglobinuria (PNH). Patient red blood cells (RBCs) are placed in mild acid; a positive result indicates PNH or congenital dyserythropoietic anemia. This is now an obsolete test for diagnosing PNH due to its low sensitivity and specificity.

CD55deficiency, also called DAF deficiency or CHAPLE syndrome, is a rare genetic disorder of the immune system. CHAPLE stands for "CD55 deficiency with hyper-activation of complement, angiopathic thrombosis, and severe protein-losing enteropathy (PLE)." The disorder usually manifests in childhood and can be life-threatening. This condition was described by Özen, et al. in 2017.
Ravulizumab, sold under the brand name Ultomiris, is a humanized monoclonal antibody complement inhibitor medication designed for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome. It is designed to bind to and prevent the activation of Complement component 5 (C5).
Pegcetacoplan, sold under the brand name Empaveli, among others, is a medication used to treat paroxysmal nocturnal hemoglobinuria and geographic atrophy of the retina. Pegcetacoplan is a complement inhibitor.

Iptacopan, sold under the brand name Fabhalta, is a medication used for the treatment of paroxysmal nocturnal hemoglobinuria. It is a complement factor B inhibitor that was developed by Novartis. It is taken by mouth.

The sucrose lysis test is a diagnostic laboratory test used for diagnosing paroxysmal nocturnal hemoglobinuria (PNH), as well as for hypoplastic anemias and any hemolytic anemia with an unclear cause. The test works by using sucrose, which creates a low ionic strength environment that allows complement to bind to red blood cells. In individuals with PNH, some red blood cells are especially vulnerable to lysis caused by complement. The test may also produce suspicious results in other hematologic conditions, including megaloblastic anemia and autoimmune hemolytic anemia. False-negative results can occur when complement activity is absent in the serum. A simpler alternative called the sugar water test also involves mixing blood with sugar and observing for hemolysis, using the same principle as the sucrose lysis test.
Crovalimab is an C5 inhibiting monoclonal antibody under investigation by Roche/Genentech for the treatment of paroxysmal nocturnal hemoglobinuria (PNH).

Danicopan, sold under the brand name Voydeya, is a medication used for the treatment of paroxysmal nocturnal hemoglobinuria. It is a complement inhibitor which reversibly binds to factor D to prevent alternative pathway-mediated hemolysis and deposition of complement C3 proteins on red blood cells.


