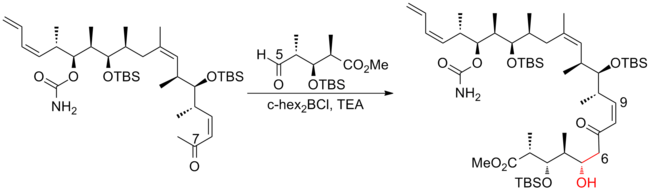
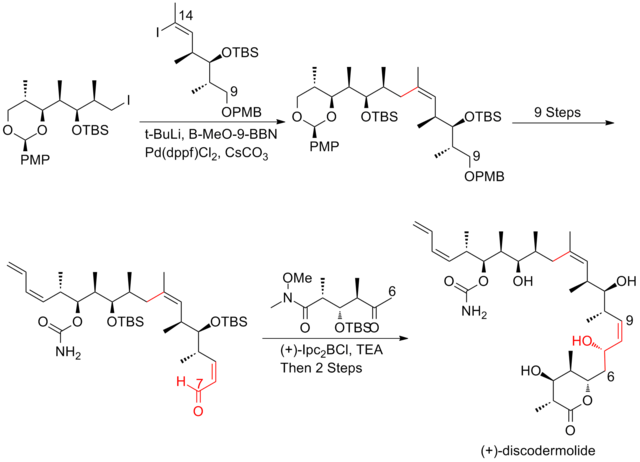
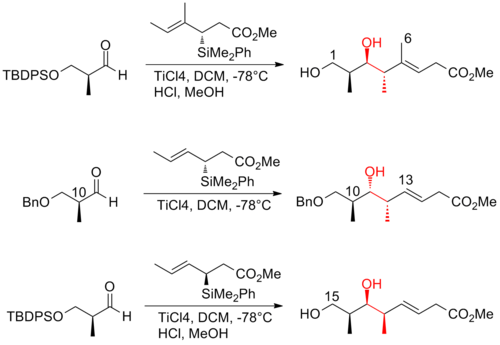
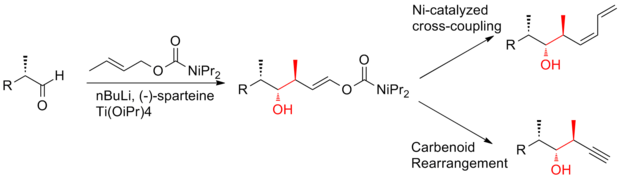
The aldol reaction is a reaction in organic chemistry that combines two carbonyl compounds to form a new β-hydroxy carbonyl compound. Its simplest form might involve the nucleophilic addition of an enolized ketone to another:

An aldol condensation is a condensation reaction in organic chemistry in which two carbonyl moieties react to form a β-hydroxyaldehyde or β-hydroxyketone, and this is then followed by dehydration to give a conjugated enone.

The Bamford–Stevens reaction is a chemical reaction whereby treatment of tosylhydrazones with strong base gives alkenes. It is named for the British chemist William Randall Bamford and the Scottish chemist Thomas Stevens Stevens (1900–2000). The usage of aprotic solvents gives predominantly Z-alkenes, while protic solvent gives a mixture of E- and Z-alkenes. As an alkene-generating transformation, the Bamford–Stevens reaction has broad utility in synthetic methodology and complex molecule synthesis.
A cascade reaction, also known as a domino reaction or tandem reaction, is a chemical process that comprises at least two consecutive reactions such that each subsequent reaction occurs only in virtue of the chemical functionality formed in the previous step. In cascade reactions, isolation of intermediates is not required, as each reaction composing the sequence occurs spontaneously. In the strictest definition of the term, the reaction conditions do not change among the consecutive steps of a cascade and no new reagents are added after the initial step. By contrast, one-pot procedures similarly allow at least two reactions to be carried out consecutively without any isolation of intermediates, but do not preclude the addition of new reagents or the change of conditions after the first reaction. Thus, any cascade reaction is also a one-pot procedure, while the reverse does not hold true. Although often composed solely of intramolecular transformations, cascade reactions can also occur intermolecularly, in which case they also fall under the category of multicomponent reactions.
The Reformatsky reaction is an organic reaction which condenses aldehydes or ketones with α-halo esters using metallic zinc to form β-hydroxy-esters:

In stereochemistry, a chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.

The Petasis reaction is the multi-component reaction of an amine, a carbonyl, and a vinyl- or aryl-boronic acid to form substituted amines.
The Stetter reaction is a reaction used in organic chemistry to form carbon-carbon bonds through a 1,4-addition reaction utilizing a nucleophilic catalyst. While the related 1,2-addition reaction, the benzoin condensation, was known since the 1830s, the Stetter reaction was not reported until 1973 by Dr. Hermann Stetter. The reaction provides synthetically useful 1,4-dicarbonyl compounds and related derivatives from aldehydes and Michael acceptors. Unlike 1,3-dicarbonyls, which are easily accessed through the Claisen condensation, or 1,5-dicarbonyls, which are commonly made using a Michael reaction, 1,4-dicarbonyls are challenging substrates to synthesize, yet are valuable starting materials for several organic transformations, including the Paal–Knorr synthesis of furans and pyrroles. Traditionally utilized catalysts for the Stetter reaction are thiazolium salts and cyanide anion, but more recent work toward the asymmetric Stetter reaction has found triazolium salts to be effective. The Stetter reaction is an example of umpolung chemistry, as the inherent polarity of the aldehyde is reversed by the addition of the catalyst to the aldehyde, rendering the carbon center nucleophilic rather than electrophilic.

Epothilones are a class of potential cancer drugs. Like taxanes, they prevent cancer cells from dividing by interfering with tubulin, but in early trials, epothilones have better efficacy and milder adverse effects than taxanes.

Organozinc chemistry is the study of the physical properties, synthesis, and reactions of organozinc compounds, which are organometallic compounds that contain carbon (C) to zinc (Zn) chemical bonds.

Zincke aldehydes, or 5-aminopenta-2,4-dienals, are the product of the reaction of a pyridinium salt with two equivalents of any secondary amine, followed by basic hydrolysis. Using secondary amines the Zincke reaction takes on a different shape forming Zincke aldehydes in which the pyridine ring is ring-opened with the terminal iminium group hydrolyzed to an aldehyde. The use of the dinitrophenyl group for pyridine activation was first reported by Theodor Zincke. The use of cyanogen bromide for pyridine activation was independently reported by W. König:
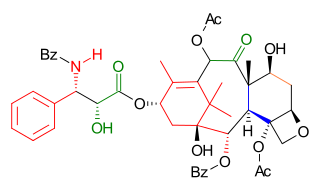
Wender Taxol total synthesis in organic chemistry describes a Taxol total synthesis by the group of Paul Wender at Stanford University published in 1997. This synthesis has much in common with the Holton Taxol total synthesis in that it is a linear synthesis starting from a naturally occurring compound with ring construction in the order A,B,C,D. The Wender effort is shorter by approximately 10 steps.

The Mukaiyama taxol total synthesis published by the group of Teruaki Mukaiyama of the Tokyo University of Science between 1997 and 1999 was the 6th successful taxol total synthesis. The total synthesis of Taxol is considered a hallmark in organic synthesis.
The Fukuyama coupling is a coupling reaction taking place between a thioester and an organozinc halide in the presence of a palladium catalyst. The reaction product is a ketone. This reaction was discovered by Tohru Fukuyama et al. in 1998.

Strychnine total synthesis in chemistry describes the total synthesis of the complex biomolecule strychnine. The first reported method by the group of Robert Burns Woodward in 1954 is considered a classic in this research field.
The Evans–Tishchenko reaction is the diastereoselective reduction of β-hydroxy ketones to the corresponding 1,3-anti diol monoesters. The reaction employs a Lewis acid, often samarium iodide, and an aldehyde. It was first described in 1990 by David A. Evans and Amir Hoveyda, as a development of the well-known Tishchenko reaction discovered in 1906. The Aldol–Tishchenko reaction provides another potential route to 1,3-diol monoester products.
William Clark Still is an American organic chemist. As a distinguished professor at Columbia University, Clark Still made significant contributions to the field of organic chemistry, particularly in the areas of natural product synthesis, reaction development, conformational analysis, macrocyclic stereocontrol, and computational chemistry. Still and coworkers also developed the purification technique known as flash column chromatography which is widely used for the purification of organic compounds.
The Kröhnke pyridine synthesis is reaction in organic synthesis between α-pyridinium methyl ketone salts and α, β-unsaturated carbonyl compounds used to generate highly functionalized pyridines. Pyridines occur widely in natural and synthetic products, so there is wide interest in routes for their synthesis. The method is named after Fritz Kröhnke.
The Crabbé reaction is an organic reaction that converts a terminal alkyne and aldehyde into an allene in the presence of a soft Lewis acid catalyst and secondary amine. Given continued developments in scope and generality, it is a convenient and increasingly important method for the preparation of allenes, a class of compounds often viewed as exotic and synthetically challenging to access.

Teruaki Mukaiyama was a Japanese organic chemist. One of the most prolific chemists of the 20th century in the field of organic synthesis, Mukaiyama helped establish the field of organic chemistry in Japan after World War II.