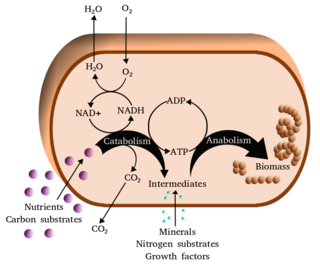
Metabolism is the set of life-sustaining chemical reactions in organisms. The three main functions of metabolism are: the conversion of the energy in food to energy available to run cellular processes; the conversion of food to building blocks of proteins, lipids, nucleic acids, and some carbohydrates; and the elimination of metabolic wastes. These enzyme-catalyzed reactions allow organisms to grow and reproduce, maintain their structures, and respond to their environments. The word metabolism can also refer to the sum of all chemical reactions that occur in living organisms, including digestion and the transportation of substances into and between different cells, in which case the above described set of reactions within the cells is called intermediary metabolism.

Molecular genetics is a branch of biology that addresses how differences in the structures or expression of DNA molecules manifests as variation among organisms. Molecular genetics often applies an "investigative approach" to determine the structure and/or function of genes in an organism's genome using genetic screens.

The metabolome refers to the complete set of small-molecule chemicals found within a biological sample. The biological sample can be a cell, a cellular organelle, an organ, a tissue, a tissue extract, a biofluid or an entire organism. The small molecule chemicals found in a given metabolome may include both endogenous metabolites that are naturally produced by an organism as well as exogenous chemicals that are not naturally produced by an organism.
A biochemical cascade, also known as a signaling cascade or signaling pathway, is a series of chemical reactions that occur within a biological cell when initiated by a stimulus. This stimulus, known as a first messenger, acts on a receptor that is transduced to the cell interior through second messengers which amplify the signal and transfer it to effector molecules, causing the cell to respond to the initial stimulus. Most biochemical cascades are series of events, in which one event triggers the next, in a linear fashion. At each step of the signaling cascade, various controlling factors are involved to regulate cellular actions, in order to respond effectively to cues about their changing internal and external environments.
BRENDA is an information system representing one of the most comprehensive enzyme repositories. It is an electronic resource that comprises molecular and biochemical information on enzymes that have been classified by the IUBMB. Every classified enzyme is characterized with respect to its catalyzed biochemical reaction. Kinetic properties of the corresponding reactants are described in detail. BRENDA contains enzyme-specific data manually extracted from primary scientific literature and additional data derived from automatic information retrieval methods such as text mining. It provides a web-based user interface that allows a convenient and sophisticated access to the data.
Metabolic network modelling, also known as metabolic network reconstruction or metabolic pathway analysis, allows for an in-depth insight into the molecular mechanisms of a particular organism. In particular, these models correlate the genome with molecular physiology. A reconstruction breaks down metabolic pathways into their respective reactions and enzymes, and analyzes them within the perspective of the entire network. In simplified terms, a reconstruction collects all of the relevant metabolic information of an organism and compiles it in a mathematical model. Validation and analysis of reconstructions can allow identification of key features of metabolism such as growth yield, resource distribution, network robustness, and gene essentiality. This knowledge can then be applied to create novel biotechnology.
Reactome is a free online database of biological pathways. There are several Reactomes that concentrate on specific organisms, the largest of these is focused on human biology, the following description concentrates on the human Reactome. It is authored by biologists, in collaboration with Reactome editorial staff. The content is cross-referenced to many bioinformatics databases. The rationale behind Reactome is to visually represent biological pathways in full mechanistic detail, while making the source data available in a computationally accessible format.
The MetaCyc database is one of the largest metabolic pathways and enzymes databases currently available. The data in the database is manually curated from the scientific literature, and covers all domains of life. MetaCyc has extensive information about chemical compounds, reactions, metabolic pathways and enzymes. The data have been curated from more than 58,000 publications.
The BioCyc database collection is an assortment of organism specific Pathway/Genome Databases (PGDBs) that provide reference to genome and metabolic pathway information for thousands of organisms. As of July 2023, there were over 20,040 databases within BioCyc. SRI International, based in Menlo Park, California, maintains the BioCyc database family.

N-Acetylserotonin O-methyltransferase, also known as ASMT, is an enzyme which catalyzes the final reaction in melatonin biosynthesis: converting Normelatonin to melatonin. This reaction is embedded in the more general tryptophan metabolism pathway. The enzyme also catalyzes a second reaction in tryptophan metabolism: the conversion of 5-hydroxy-indoleacetate to 5-methoxy-indoleacetate. The other enzyme which catalyzes this reaction is n-acetylserotonin-o-methyltransferase-like-protein.
Microbial biodegradation is the use of bioremediation and biotransformation methods to harness the naturally occurring ability of microbial xenobiotic metabolism to degrade, transform or accumulate environmental pollutants, including hydrocarbons, polychlorinated biphenyls (PCBs), polyaromatic hydrocarbons (PAHs), heterocyclic compounds, pharmaceutical substances, radionuclides and metals.

MicrobesOnline is a publicly and freely accessible website that hosts multiple comparative genomic tools for comparing microbial species at the genomic, transcriptomic and functional levels. MicrobesOnline was developed by the Virtual Institute for Microbial Stress and Survival, which is based at the Lawrence Berkeley National Laboratory in Berkeley, California. The site was launched in 2005, with regular updates until 2011.

In molecular biology, STRING is a biological database and web resource of known and predicted protein–protein interactions.
The Small Molecule Pathway Database (SMPDB) is a comprehensive, high-quality, freely accessible, online database containing more than 600 small molecule (i.e. metabolic) pathways found in humans. SMPDB is designed specifically to support pathway elucidation and pathway discovery in metabolomics, transcriptomics, proteomics and systems biology. It is able to do so, in part, by providing colorful, detailed, fully searchable, hyperlinked diagrams of five types of small molecule pathways: 1) general human metabolic pathways; 2) human metabolic disease pathways; 3) human metabolite signaling pathways; 4) drug-action pathways and 5) drug metabolism pathways. SMPDB pathways may be navigated, viewed and zoomed interactively using a Google Maps-like interface. All SMPDB pathways include information on the relevant organs, subcellular compartments, protein cofactors, protein locations, metabolite locations, chemical structures and protein quaternary structures (Fig. 1). Each small molecule in SMPDB is hyperlinked to detailed descriptions contained in the HMDB or DrugBank and each protein or enzyme complex is hyperlinked to UniProt. Additionally, all SMPDB pathways are accompanied with detailed descriptions and references, providing an overview of the pathway, condition or processes depicted in each diagram. Users can browse the SMPDB (Fig. 2) or search its contents by text searching (Fig. 3), sequence searching, or chemical structure searching. More powerful queries are also possible including searching with lists of gene or protein names, drug names, metabolite names, GenBank IDs, Swiss-Prot IDs, Agilent or Affymetrix microarray IDs. These queries will produce lists of matching pathways and highlight the matching molecules on each of the pathway diagrams. Gene, metabolite and protein concentration data can also be visualized through SMPDB's mapping interface.
Metabolomic Pathway Analysis, shortened to MetPA, is a freely available, user-friendly web server to assist with the identification analysis and visualization of metabolic pathways using metabolomic data. MetPA makes use of advances originally developed for pathway analysis in microarray experiments and applies those principles and concepts to the analysis of metabolic pathways. For input, MetPA expects either a list of compound names or a metabolite concentration table with phenotypic labels. The list of compounds can include common names, HMDB IDs or KEGG IDs with one compound per row. Compound concentration tables must have samples in rows and compounds in columns. MetPA's output is a series of tables indicating which pathways are significantly enriched as well as a variety of graphs or pathway maps illustrating where and how certain pathways were enriched. MetPA's graphical output uses a colorful Google-Maps visualization system that allows simple, intuitive data exploration that lets users employ a computer mouse or track pad to select, drag and place images and to seamlessly zoom in and out. Users can explore MetPA's output using three different views or levels: 1) a metabolome view; 2) a pathway view; 3) a compound view.
In bioinformatics, a Gene Disease Database is a systematized collection of data, typically structured to model aspects of reality, in a way to comprehend the underlying mechanisms of complex diseases, by understanding multiple composite interactions between phenotype-genotype relationships and gene-disease mechanisms. Gene Disease Databases integrate human gene-disease associations from various expert curated databases and text mining derived associations including Mendelian, complex and environmental diseases.
Model organism databases (MODs) are biological databases, or knowledgebases, dedicated to the provision of in-depth biological data for intensively studied model organisms. MODs allow researchers to easily find background information on large sets of genes, plan experiments efficiently, combine their data with existing knowledge, and construct novel hypotheses. They allow users to analyse results and interpret datasets, and the data they generate are increasingly used to describe less well studied species. Where possible, MODs share common approaches to collect and represent biological information. For example, all MODs use the Gene Ontology (GO) to describe functions, processes and cellular locations of specific gene products. Projects also exist to enable software sharing for curation, visualization and querying between different MODs. Organismal diversity and varying user requirements however mean that MODs are often required to customize capture, display, and provision of data.
Minoru Kanehisa is a Japanese bioinformatician. He is a project professor at Kyoto University, technical director of Pathway Solutions Inc and president of NPO Bioinformatics Japan. He is one of Japan's most recognized and respected bioinformatics experts and is known for developing the KEGG bioinformatics database.
Genome mining describes the exploitation of genomic information for the discovery of biosynthetic pathways of natural products and their possible interactions. It depends on computational technology and bioinformatics tools. The mining process relies on a huge amount of data accessible in genomic databases. By applying data mining algorithms, the data can be used to generate new knowledge in several areas of medicinal chemistry, such as discovering novel natural products.