
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences, such as calculating the distance cost between strings in a natural language or in financial data.
UPGMA is a simple agglomerative (bottom-up) hierarchical clustering method. It also has a weighted variant, WPGMA, and they are generally attributed to Sokal and Michener.
In mathematics, computer science and especially graph theory, a distance matrix is a square matrix containing the distances, taken pairwise, between the elements of a set. Depending upon the application involved, the distance being used to define this matrix may or may not be a metric. If there are N elements, this matrix will have size N×N. In graph-theoretic applications, the elements are more often referred to as points, nodes or vertices.
FASTA is a DNA and protein sequence alignment software package first described by David J. Lipman and William R. Pearson in 1985. Its legacy is the FASTA format which is now ubiquitous in bioinformatics.

The Smith–Waterman algorithm performs local sequence alignment; that is, for determining similar regions between two strings of nucleic acid sequences or protein sequences. Instead of looking at the entire sequence, the Smith–Waterman algorithm compares segments of all possible lengths and optimizes the similarity measure.
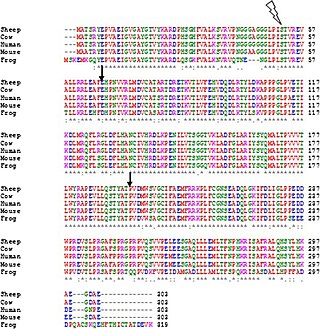
Clustal is a series of widely used computer programs used in bioinformatics for multiple sequence alignment. There have been many versions of Clustal over the development of the algorithm that are listed below. The analysis of each tool and its algorithm is also detailed in their respective categories. Available operating systems listed in the sidebar are a combination of the software availability and may not be supported for every current version of the Clustal tools. Clustal Omega has the widest variety of operating systems out of all the Clustal tools.
The European Bioinformatics Institute (EMBL-EBI) is an intergovernmental organization (IGO) which, as part of the European Molecular Biology Laboratory (EMBL) family, focuses on research and services in bioinformatics. It is located on the Wellcome Genome Campus in Hinxton near Cambridge, and employs over 600 full-time equivalent (FTE) staff. Institute leaders such as Rolf Apweiler, Alex Bateman, Ewan Birney, and Guy Cochrane, an adviser on the National Genomics Data Center Scientific Advisory Board, serve as part of the international research network of the BIG Data Center at the Beijing Institute of Genomics.
In computational phylogenetics, tree alignment is a computational problem concerned with producing multiple sequence alignments, or alignments of three or more sequences of DNA, RNA, or protein. Sequences are arranged into a phylogenetic tree, modeling the evolutionary relationships between species or taxa. The edit distances between sequences are calculated for each of the tree's internal vertices, such that the sum of all edit distances within the tree is minimized. Tree alignment can be accomplished using one of several algorithms with various trade-offs between manageable tree size and computational effort.
Computational phylogenetics, phylogeny inference, or phylogenetic inference focuses on computational and optimization algorithms, heuristics, and approaches involved in phylogenetic analyses. The goal is to find a phylogenetic tree representing optimal evolutionary ancestry between a set of genes, species, or taxa. Maximum likelihood, parsimony, Bayesian, and minimum evolution are typical optimality criteria used to assess how well a phylogenetic tree topology describes the sequence data. Nearest Neighbour Interchange (NNI), Subtree Prune and Regraft (SPR), and Tree Bisection and Reconnection (TBR), known as tree rearrangements, are deterministic algorithms to search for optimal or the best phylogenetic tree. The space and the landscape of searching for the optimal phylogenetic tree is known as phylogeny search space.

Multiple sequence alignment (MSA) may refer to the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a linkage and are descended from a common ancestor. From the resulting MSA, sequence homology can be inferred and phylogenetic analysis can be conducted to assess the sequences' shared evolutionary origins. Visual depictions of the alignment as in the image at right illustrate mutation events such as point mutations that appear as differing characters in a single alignment column, and insertion or deletion mutations that appear as hyphens in one or more of the sequences in the alignment. Multiple sequence alignment is often used to assess sequence conservation of protein domains, tertiary and secondary structures, and even individual amino acids or nucleotides.
InterPro is a database of protein families, protein domains and functional sites in which identifiable features found in known proteins can be applied to new protein sequences in order to functionally characterise them.
T-Coffee is a multiple sequence alignment software using a progressive approach. It generates a library of pairwise alignments to guide the multiple sequence alignment. It can also combine multiple sequences alignments obtained previously and in the latest versions can use structural information from PDB files (3D-Coffee). It has advanced features to evaluate the quality of the alignments and some capacity for identifying occurrence of motifs (Mocca). It produces alignment in the aln format (Clustal) by default, but can also produce PIR, MSF, and FASTA format. The most common input formats are supported.
In bioinformatics, MAFFT is a program used to create multiple sequence alignments of amino acid or nucleotide sequences. Published in 2002, the first version of MAFFT used an algorithm based on progressive alignment, in which the sequences were clustered with the help of the Fast Fourier Transform. Subsequent versions of MAFFT have added other algorithms and modes of operation, including options for faster alignment of large numbers of sequences, higher accuracy alignments, alignment of non-coding RNA sequences, and the addition of new sequences to existing alignments.
Distance matrices are used in phylogeny as non-parametric distance methods and were originally applied to phenetic data using a matrix of pairwise distances. These distances are then reconciled to produce a tree. The distance matrix can come from a number of different sources, including measured distance or morphometric analysis, various pairwise distance formulae applied to discrete morphological characters, or genetic distance from sequence, restriction fragment, or allozyme data. For phylogenetic character data, raw distance values can be calculated by simply counting the number of pairwise differences in character states.

UGENE is computer software for bioinformatics. It works on personal computer operating systems such as Windows, macOS, or Linux. It is released as free and open-source software, under a GNU General Public License (GPL) version 2.

HMMER is a free and commonly used software package for sequence analysis written by Sean Eddy. Its general usage is to identify homologous protein or nucleotide sequences, and to perform sequence alignments. It detects homology by comparing a profile-HMM to either a single sequence or a database of sequences. Sequences that score significantly better to the profile-HMM compared to a null model are considered to be homologous to the sequences that were used to construct the profile-HMM. Profile-HMMs are constructed from a multiple sequence alignment in the HMMER package using the hmmbuild program. The profile-HMM implementation used in the HMMER software was based on the work of Krogh and colleagues. HMMER is a console utility ported to every major operating system, including different versions of Linux, Windows, and macOS.

Molecular Evolutionary Genetics Analysis (MEGA) is computer software for conducting statistical analysis of molecular evolution and for constructing phylogenetic trees. It includes many sophisticated methods and tools for phylogenomics and phylomedicine. It is licensed as proprietary freeware. The project for developing this software was initiated by the leadership of Masatoshi Nei in his laboratory at the Pennsylvania State University in collaboration with his graduate student Sudhir Kumar and postdoctoral fellow Koichiro Tamura. Nei wrote a monograph (pp. 130) outlining the scope of the software and presenting new statistical methods that were included in MEGA. The entire set of computer programs was written by Kumar and Tamura. The personal computers then lacked the ability to send the monograph and software electronically, so they were delivered by postal mail. From the start, MEGA was intended to be easy-to-use and include solid statistical methods only.
T-REX is a freely available web server, developed at the department of Computer Science of the Université du Québec à Montréal, dedicated to the inference, validation and visualization of phylogenetic trees and phylogenetic networks. The T-REX web server allows the users to perform several popular methods of phylogenetic analysis as well as some new phylogenetic applications for inferring, drawing and validating phylogenetic trees and networks.
In bioinformatics, alignment-free sequence analysis approaches to molecular sequence and structure data provide alternatives over alignment-based approaches.

Desmond Gerard Higgins is a Professor of Bioinformatics at University College Dublin, widely known for CLUSTAL, a series of computer programs for performing multiple sequence alignment. According to Nature, Higgins' papers describing CLUSTAL are among the top ten most highly cited scientific papers of all time.
