
Portable Document Format (PDF), standardized as ISO 32000, is a file format developed by Adobe in 1992 to present documents, including text formatting and images, in a manner independent of application software, hardware, and operating systems. Based on the PostScript language, each PDF file encapsulates a complete description of a fixed-layout flat document, including the text, fonts, vector graphics, raster images and other information needed to display it. PDF has its roots in "The Camelot Project" initiated by Adobe co-founder John Warnock in 1991. PDF was standardized as ISO 32000 in 2008. The last edition as ISO 32000-2:2020 was published in December 2020.
Waveform Audio File Format is an audio file format standard, developed by IBM and Microsoft, for storing an audio bitstream on personal computers. It is the main format used on Microsoft Windows systems for uncompressed audio. The usual bitstream encoding is the linear pulse-code modulation (LPCM) format.
Resource Interchange File Format (RIFF) is a generic file container format for storing data in tagged chunks. It is primarily used for audio and video, though it can be used for arbitrary data.
ZIP is an archive file format that supports lossless data compression. A ZIP file may contain one or more files or directories that may have been compressed. The ZIP file format permits a number of compression algorithms, though DEFLATE is the most common. This format was originally created in 1989 and was first implemented in PKWARE, Inc.'s PKZIP utility, as a replacement for the previous ARC compression format by Thom Henderson. The ZIP format was then quickly supported by many software utilities other than PKZIP. Microsoft has included built-in ZIP support in versions of Microsoft Windows since 1998 via the "Plus! 98" addon for Windows 98. Native support was added as of the year 2000 in Windows ME. Apple has included built-in ZIP support in Mac OS X 10.3 and later. Most free operating systems have built in support for ZIP in similar manners to Windows and Mac OS X.
In bioinformatics and biochemistry, the FASTA format is a text-based format for representing either nucleotide sequences or amino acid (protein) sequences, in which nucleotides or amino acids are represented using single-letter codes.
The archiver, also known simply as ar, is a Unix utility that maintains groups of files as a single archive file. Today, ar is generally used only to create and update static library files that the link editor or linker uses and for generating .deb packages for the Debian family; it can be used to create archives for any purpose, but has been largely replaced by tar for purposes other than static libraries. An implementation of ar is included as one of the GNU Binutils.

Comma-separated values (CSV) is a text file format that uses commas to separate values. A CSV file stores tabular data in plain text, where each line of the file typically represents one data record. Each record consists of the same number of fields, and these are separated by commas in the CSV file. If the field delimiter itself may appear within a field, fields can be surrounded with quotation marks.
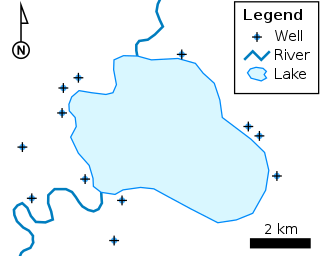
The shapefile format is a geospatial vector data format for geographic information system (GIS) software. It is developed and regulated by Esri as a mostly open specification for data interoperability among Esri and other GIS software products. The shapefile format can spatially describe vector features: points, lines, and polygons, representing, for example, water wells, rivers, and lakes. Each item usually has attributes that describe it, such as name or temperature.
Real-Time Messaging Protocol (RTMP) is a communication protocol for streaming audio, video, and data over the Internet. Originally developed as a proprietary protocol by Macromedia for streaming between Flash Player and the Flash Communication Server, Adobe has released an incomplete version of the specification of the protocol for public use.
FASTQ format is a text-based format for storing both a biological sequence and its corresponding quality scores. Both the sequence letter and quality score are each encoded with a single ASCII character for brevity.
Pileup format is a text-based format for summarizing the base calls of aligned reads to a reference sequence. This format facilitates visual display of SNP/indel calling and alignment. It was first used by Tony Cox and Zemin Ning at the Wellcome Trust Sanger Institute, and became widely known through its implementation within the SAMtools software suite.
SAMtools is a set of utilities for interacting with and post-processing short DNA sequence read alignments in the SAM, BAM and CRAM formats, written by Heng Li. These files are generated as output by short read aligners like BWA. Both simple and advanced tools are provided, supporting complex tasks like variant calling and alignment viewing as well as sorting, indexing, data extraction and format conversion. SAM files can be very large, so compression is used to save space. SAM files are human-readable text files, and BAM files are simply their binary equivalent, whilst CRAM files are a restructured column-oriented binary container format. BAM files are typically compressed and more efficient for software to work with than SAM. SAMtools makes it possible to work directly with a compressed BAM file, without having to uncompress the whole file. Additionally, since the format for a SAM/BAM file is somewhat complex - containing reads, references, alignments, quality information, and user-specified annotations - SAMtools reduces the effort needed to use SAM/BAM files by hiding low-level details.
Ensembl Genomes is a scientific project to provide genome-scale data from non-vertebrate species.
GeneTalk is a web-based platform, tool, and database for filtering, reduction and prioritization of human sequence variants from next-generation sequencing (NGS) data. GeneTalk allows editing annotation about sequence variants and build up a crowd sourced database with clinically relevant information for diagnostics of genetic disorders. GeneTalk allows searching for information about specific sequence variants and connects to experts on variants that are potentially disease-relevant.
Sequence Alignment Map (SAM) is a text-based format originally for storing biological sequences aligned to a reference sequence developed by Heng Li and Bob Handsaker et al. It was developed when the 1000 Genomes Project wanted to move away from the MAQ mapper format and decided to design a new format. The overall TAB-delimited flavour of the format came from an earlier format inspired by BLAT’s PSL. The name of SAM came from Gabor Marth from University of Utah, who originally had a format under the same name but with a different syntax more similar to a BLAST output. It is widely used for storing data, such as nucleotide sequences, generated by next generation sequencing technologies, and the standard has been broadened to include unmapped sequences. The format supports short and long reads (up to 128 Mbp) produced by different sequencing platforms and is used to hold mapped data within the Genome Analysis Toolkit (GATK) and across the Broad Institute, the Wellcome Sanger Institute, and throughout the 1000 Genomes Project.

Binary Alignment Map (BAM) is the comprehensive raw data of genome sequencing; it consists of the lossless, compressed binary representation of the Sequence Alignment Map-files.
Compressed Reference-oriented Alignment Map (CRAM) is a compressed columnar file format for storing biological sequences aligned to a reference sequence, initially devised by Markus Hsi-Yang Fritz et al.
ANNOVAR is a bioinformatics software tool for the interpretation and prioritization of single nucleotide variants (SNVs), insertions, deletions, and copy number variants (CNVs) of a given genome.
The BED format is a text file format used to store genomic regions as coordinates and associated annotations. The data are presented in the form of columns separated by spaces or tabs. This format was developed during the Human Genome Project and then adopted by other sequencing projects. As a result of this increasingly wide use, this format had already become a de facto standard in bioinformatics before a formal specification was written.
Tabix is a bioinformatics software utility for indexing large genomic data files. Tabix is free software under the MIT license.
