
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences such as calculating the distance cost between strings in a natural language, or to display financial data.
In bioinformatics, sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of a wide range of analytical methods to understand its features, function, structure, or evolution. Methodologies used include sequence alignment, searches against biological databases, and others.
In bioinformatics and evolutionary biology, a substitution matrix describes the frequency at which a character in a nucleotide sequence or a protein sequence changes to other character states over evolutionary time. The information is often in the form of log odds of finding two specific character states aligned and depends on the assumed number of evolutionary changes or sequence dissimilarity between compared sequences. It is an application of a stochastic matrix. Substitution matrices are usually seen in the context of amino acid or DNA sequence alignments, where they are used to calculate similarity scores between the aligned sequences.
In bioinformatics, BLAST is an algorithm and program for comparing primary biological sequence information, such as the amino-acid sequences of proteins or the nucleotides of DNA and/or RNA sequences. A BLAST search enables a researcher to compare a subject protein or nucleotide sequence with a library or database of sequences, and identify database sequences that resemble the query sequence above a certain threshold. For example, following the discovery of a previously unknown gene in the mouse, a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene; BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence.
FASTA is a DNA and protein sequence alignment software package first described by David J. Lipman and William R. Pearson in 1985. Its legacy is the FASTA format which is now ubiquitous in bioinformatics.

In bioinformatics, a sequence logo is a graphical representation of the sequence conservation of nucleotides or amino acids . A sequence logo is created from a collection of aligned sequences and depicts the consensus sequence and diversity of the sequences. Sequence logos are frequently used to depict sequence characteristics such as protein-binding sites in DNA or functional units in proteins.

The Smith–Waterman algorithm performs local sequence alignment; that is, for determining similar regions between two strings of nucleic acid sequences or protein sequences. Instead of looking at the entire sequence, the Smith–Waterman algorithm compares segments of all possible lengths and optimizes the similarity measure.
The European Bioinformatics Institute (EMBL-EBI) is an intergovernmental organization (IGO) which, as part of the European Molecular Biology Laboratory (EMBL) family, focuses on research and services in bioinformatics. It is located on the Wellcome Genome Campus in Hinxton near Cambridge, and employs over 600 full-time equivalent (FTE) staff. Institute leaders such as Rolf Apweiler, Alex Bateman, Ewan Birney, and Guy Cochrane, an adviser on the National Genomics Data Center Scientific Advisory Board, serve as part of the international research network of the BIG Data Center at the Beijing Institute of Genomics.

Pfam is a database of protein families that includes their annotations and multiple sequence alignments generated using hidden Markov models. The most recent version, Pfam 36.0, was released in September 2023 and contains 20,795 families.

Multiple sequence alignment (MSA) may refer to the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a linkage and are descended from a common ancestor. From the resulting MSA, sequence homology can be inferred and phylogenetic analysis can be conducted to assess the sequences' shared evolutionary origins. Visual depictions of the alignment as in the image at right illustrate mutation events such as point mutations that appear as differing characters in a single alignment column, and insertion or deletion mutations that appear as hyphens in one or more of the sequences in the alignment. Multiple sequence alignment is often used to assess sequence conservation of protein domains, tertiary and secondary structures, and even individual amino acids or nucleotides.
InterPro is a database of protein families, protein domains and functional sites in which identifiable features found in known proteins can be applied to new protein sequences in order to functionally characterise them.
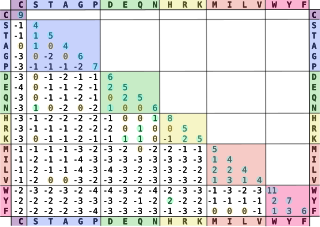
In bioinformatics, the BLOSUM matrix is a substitution matrix used for sequence alignment of proteins. BLOSUM matrices are used to score alignments between evolutionarily divergent protein sequences. They are based on local alignments. BLOSUM matrices were first introduced in a paper by Steven Henikoff and Jorja Henikoff. They scanned the BLOCKS database for very conserved regions of protein families and then counted the relative frequencies of amino acids and their substitution probabilities. Then, they calculated a log-odds score for each of the 210 possible substitution pairs of the 20 standard amino acids. All BLOSUM matrices are based on observed alignments; they are not extrapolated from comparisons of closely related proteins like the PAM Matrices.
BLAT is a pairwise sequence alignment algorithm that was developed by Jim Kent at the University of California Santa Cruz (UCSC) in the early 2000s to assist in the assembly and annotation of the human genome. It was designed primarily to decrease the time needed to align millions of mouse genomic reads and expressed sequence tags against the human genome sequence. The alignment tools of the time were not capable of performing these operations in a manner that would allow a regular update of the human genome assembly. Compared to pre-existing tools, BLAT was ~500 times faster with performing mRNA/DNA alignments and ~50 times faster with protein/protein alignments.
CS-BLAST (Context-Specific BLAST) is a tool that searches a protein sequence that extends BLAST, using context-specific mutation probabilities. More specifically, CS-BLAST derives context-specific amino-acid similarities on each query sequence from short windows on the query sequences. Using CS-BLAST doubles sensitivity and significantly improves alignment quality without a loss of speed in comparison to BLAST. CSI-BLAST is the context-specific analog of PSI-BLAST, which computes the mutation profile with substitution probabilities and mixes it with the query profile. CSI-BLAST is the context specific analog of PSI-BLAST. Both of these programs are available as web-server and are available for free download.
SUPERFAMILY is a database and search platform of structural and functional annotation for all proteins and genomes. It classifies amino acid sequences into known structural domains, especially into SCOP superfamilies. Domains are functional, structural, and evolutionary units that form proteins. Domains of common Ancestry are grouped into superfamilies. The domains and domain superfamilies are defined and described in SCOP. Superfamilies are groups of proteins which have structural evidence to support a common evolutionary ancestor but may not have detectable sequence homology.
FSA is a multiple sequence alignment program for aligning many proteins or RNAs or long genomic DNA sequences. Along with MUSCLE and MAFFT, FSA is one of the few sequence alignment programs which can align datasets of hundreds or thousands of sequences. FSA uses a different optimization criterion which allows it to more reliably identify non-homologous sequences than these other programs, although this increased accuracy comes at the cost of decreased speed.
Phyre and Phyre2 are free web-based services for protein structure prediction. Phyre is among the most popular methods for protein structure prediction having been cited over 1500 times. Like other remote homology recognition techniques, it is able to regularly generate reliable protein models when other widely used methods such as PSI-BLAST cannot. Phyre2 has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods. Its development is funded by the Biotechnology and Biological Sciences Research Council.

Sean Roberts Eddy is Professor of Molecular & Cellular Biology and of Applied Mathematics at Harvard University. Previously he was based at the Janelia Research Campus from 2006 to 2015 in Virginia. His research interests are in bioinformatics, computational biology and biological sequence analysis. As of 2016 projects include the use of Hidden Markov models in HMMER, Infernal Pfam and Rfam.
The HH-suite is an open-source software package for sensitive protein sequence searching. It contains programs that can search for similar protein sequences in protein sequence databases. Sequence searches are a standard tool in modern biology with which the function of unknown proteins can be inferred from the functions of proteins with similar sequences. HHsearch and HHblits are two main programs in the package and the entry point to its search function, the latter being a faster iteration. HHpred is an online server for protein structure prediction that uses homology information from HH-suite.


