Related Research Articles
Protein engineering is the process of developing useful or valuable proteins through the design and production of unnatural polypeptides, often by altering amino acid sequences found in nature. It is a young discipline, with much research taking place into the understanding of protein folding and recognition for protein design principles. It has been used to improve the function of many enzymes for industrial catalysis. It is also a product and services market, with an estimated value of $168 billion by 2017.

A cloning vector is a small piece of DNA that can be stably maintained in an organism, and into which a foreign DNA fragment can be inserted for cloning purposes. The cloning vector may be DNA taken from a virus, the cell of a higher organism, or it may be the plasmid of a bacterium. The vector contains features that allow for the convenient insertion of a DNA fragment into the vector or its removal from the vector, for example through the presence of restriction sites. The vector and the foreign DNA may be treated with a restriction enzyme that cuts the DNA, and DNA fragments thus generated contain either blunt ends or overhangs known as sticky ends, and vector DNA and foreign DNA with compatible ends can then be joined by molecular ligation. After a DNA fragment has been cloned into a cloning vector, it may be further subcloned into another vector designed for more specific use.
Site-directed mutagenesis is a molecular biology method that is used to make specific and intentional mutating changes to the DNA sequence of a gene and any gene products. Also called site-specific mutagenesis or oligonucleotide-directed mutagenesis, it is used for investigating the structure and biological activity of DNA, RNA, and protein molecules, and for protein engineering.
This is a list of topics in molecular biology. See also index of biochemistry articles.
A DNA construct is an artificially-designed segment of DNA borne on a vector that can be used to incorporate genetic material into a target tissue or cell. A DNA construct contains a DNA insert, called a transgene, delivered via a transformation vector which allows the insert sequence to be replicated and/or expressed in the target cell. This gene can be cloned from a naturally occurring gene, or synthetically constructed. The vector can be delivered using physical, chemical or viral methods. Typically, the vectors used in DNA constructs contain an origin of replication, a multiple cloning site, and a selectable marker. Certain vectors can carry additional regulatory elements based on the expression system involved.
A restriction digest is a procedure used in molecular biology to prepare DNA for analysis or other processing. It is sometimes termed DNA fragmentation, though this term is used for other procedures as well. In a restriction digest, DNA molecules are cleaved at specific restriction sites of 4-12 nucleotides in length by use of restriction enzymes which recognize these sequences.

In molecular biology, subcloning is a technique used to move a particular DNA sequence from a parent vector to a destination vector.
P elements are transposable elements that were discovered in Drosophila as the causative agents of genetic traits called hybrid dysgenesis. The transposon is responsible for the P trait of the P element and it is found only in wild flies. They are also found in many other eukaryotes.
The GFP-cDNA project documents the localisation of proteins to subcellular compartments of the eukaryotic cell applying fluorescence microscopy. Experimental data are complemented with bioinformatic analyses and published online in a database. A search function allows the finding of proteins containing features or motifs of particular interest. The project is a collaboration of the research groups of Rainer Pepperkok at the European Molecular Biology Laboratory (EMBL) and Stefan Wiemann at the German Cancer Research Centre (DKFZ).
Cre-Lox recombination is a site-specific recombinase technology, used to carry out deletions, insertions, translocations and inversions at specific sites in the DNA of cells. It allows the DNA modification to be targeted to a specific cell type or be triggered by a specific external stimulus. It is implemented both in eukaryotic and prokaryotic systems. The Cre-lox recombination system has been particularly useful to help neuroscientists to study the brain in which complex cell types and neural circuits come together to generate cognition and behaviors. NIH Blueprint for Neuroscience Research has created several hundreds of Cre driver mouse lines which are currently used by the worldwide neuroscience community.
Site-specific recombinase technologies are genome engineering tools that depend on recombinase enzymes to replace targeted sections of DNA.
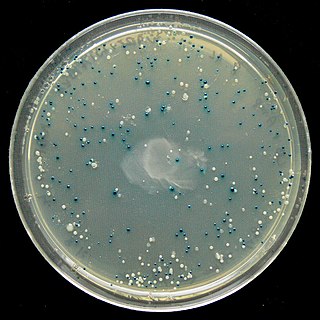
The blue–white screen is a screening technique that allows for the rapid and convenient detection of recombinant bacteria in vector-based molecular cloning experiments. This method of screening is usually performed using a suitable bacterial strain, but other organisms such as yeast may also be used. DNA of transformation is ligated into a vector. The vector is then inserted into a competent host cell viable for transformation, which are then grown in the presence of X-gal. Cells transformed with vectors containing recombinant DNA will produce white colonies; cells transformed with non-recombinant plasmids grow into blue colonies.
P1 is a temperate bacteriophage that infects Escherichia coli and some other bacteria. When undergoing a lysogenic cycle the phage genome exists as a plasmid in the bacterium unlike other phages that integrate into the host DNA. P1 has an icosahedral head containing the DNA attached to a contractile tail with six tail fibers. The P1 phage has gained research interest because it can be used to transfer DNA from one bacterial cell to another in a process known as transduction. As it replicates during its lytic cycle it captures fragments of the host chromosome. If the resulting viral particles are used to infect a different host the captured DNA fragments can be integrated into the new host's genome. This method of in vivo genetic engineering was widely used for many years and is still used today, though to a lesser extent. P1 can also be used to create the P1-derived artificial chromosome cloning vector which can carry relatively large fragments of DNA. P1 encodes a site-specific recombinase, Cre, that is widely used to carry out cell-specific or time-specific DNA recombination by flanking the target DNA with loxP sites.
Artificial gene synthesis, or simply gene synthesis, refers to a group of methods that are used in synthetic biology to construct and assemble genes from nucleotides de novo. Unlike DNA synthesis in living cells, artificial gene synthesis does not require template DNA, allowing virtually any DNA sequence to be synthesized in the laboratory. It comprises two main steps, the first of which is solid-phase DNA synthesis, sometimes known as DNA printing. This produces oligonucleotide fragments that are generally under 200 base pairs. The second step then involves connecting these oligonucleotide fragments using various DNA assembly methods. Because artificial gene synthesis does not require template DNA, it is theoretically possible to make a completely synthetic DNA molecule with no limits on the nucleotide sequence or size.

Functional cloning is a molecular cloning technique that relies on prior knowledge of the encoded protein’s sequence or function for gene identification. In this assay, a genomic or cDNA library is screened to identify the genetic sequence of a protein of interest. Expression cDNA libraries may be screened with antibodies specific for the protein of interest or may rely on selection via the protein function. Historically, the amino acid sequence of a protein was used to prepare degenerate oligonucleotides which were then probed against the library to identify the gene encoding the protein of interest. Once candidate clones carrying the gene of interest are identified, they are sequenced and their identity is confirmed. This method of cloning allows researchers to screen entire genomes without prior knowledge of the location of the gene or the genetic sequence.
In molecular cloning, a vector is any particle used as a vehicle to artificially carry a foreign nucleic sequence – usually DNA – into another cell, where it can be replicated and/or expressed. A vector containing foreign DNA is termed recombinant DNA. The four major types of vectors are plasmids, viral vectors, cosmids, and artificial chromosomes. Of these, the most commonly used vectors are plasmids. Common to all engineered vectors are an origin of replication, a multicloning site, and a selectable marker.

Molecular cloning is a set of experimental methods in molecular biology that are used to assemble recombinant DNA molecules and to direct their replication within host organisms. The use of the word cloning refers to the fact that the method involves the replication of one molecule to produce a population of cells with identical DNA molecules. Molecular cloning generally uses DNA sequences from two different organisms: the species that is the source of the DNA to be cloned, and the species that will serve as the living host for replication of the recombinant DNA. Molecular cloning methods are central to many contemporary areas of modern biology and medicine.
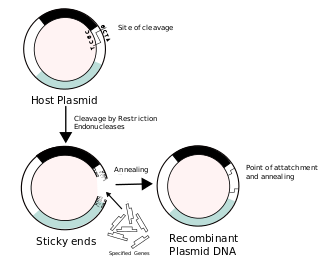
Recombinant DNA (rDNA), or molecular cloning, is the process by which a single gene, or segment of DNA, is isolated and amplified. Recombinant DNA is also known as in vitro recombination. A cloning vector is a DNA molecule that carries foreign DNA into a host cell, where it replicates, producing many copies of itself along with the foreign DNA. There are many types of cloning vectors such as plasmids and phages. In order to carry out recombination between vector and the foreign DNA, it is necessary the vector and DNA to be cloned by digestion, ligase the foreign DNA into the vector with the enzyme DNA ligase. And DNA is inserted by introducing the DNA into bacteria cells by transformation.

Ligation is the joining of two nucleic acid fragments through the action of an enzyme. It is an essential laboratory procedure in the molecular cloning of DNA, whereby DNA fragments are joined to create recombinant DNA molecules (such as when a foreign DNA fragment is inserted into a plasmid). The ends of DNA fragments are joined by the formation of phosphodiester bonds between the 3'-hydroxyl of one DNA terminus with the 5'-phosphoryl of another. RNA may also be ligated similarly. A co-factor is generally involved in the reaction, and this is usually ATP or NAD+. Eukaryotic cells ligases belong to ATP type, and NAD+ - dependent are found in bacteria (e.g. E. coli).

Golden Gate Cloning or Golden Gate assembly is a molecular cloning method that allows a researcher to simultaneously and directionally assemble multiple DNA fragments into a single piece using Type IIS restriction enzymes and T4 DNA ligase. This assembly is performed in vitro. Most commonly used Type IIS enzymes include BsaI, BsmBI, and BbsI.
References
- Hartley JL, Temple GF, Brasch MA (November 2000). "DNA cloning using in vitro site-specific recombination". Genome Res. 10 (11): 1788–95. doi:10.1101/gr.143000. PMC 310948 . PMID 11076863.
- Katzen F (April 2007). "Gateway(®) recombinational cloning: a biological operating system". Expert Opin Drug Discov. 2 (4): 571–89. doi:10.1517/17460441.2.4.571. PMID 23484762. S2CID 22606399.
- Freuler F, Stettler T, Meyerhofer M, Leder L, Mayr LM (June 2008). "Development of a novel Gateway-based vector system for efficient, multiparallel protein expression in Escherichia coli". Protein Expr. Purif. 59 (2): 232–41. doi:10.1016/j.pep.2008.02.003. PMID 18375142.
- Hartley JL. Use of the Gateway System for Protein Expression in Multiple Hosts. Curr Protoc Protein Sci. 2003 Feb;Chapter 5:Unit 5.17. PubMed PMID:18429245.
- Ptashne, M. (1992). A Genetic Switch: Phage (Lambda) and Higher Organisms (Cambridge, MA: Cell Press).