
In molecular biology, messenger ribonucleic acid (mRNA) is a single-stranded molecule of RNA that corresponds to the genetic sequence of a gene, and is read by a ribosome in the process of synthesizing a protein.
The coding region of a gene, also known as the coding sequence(CDS), is the portion of a gene's DNA or RNA that codes for a protein. Studying the length, composition, regulation, splicing, structures, and functions of coding regions compared to non-coding regions over different species and time periods can provide a significant amount of important information regarding gene organization and evolution of prokaryotes and eukaryotes. This can further assist in mapping the human genome and developing gene therapy.

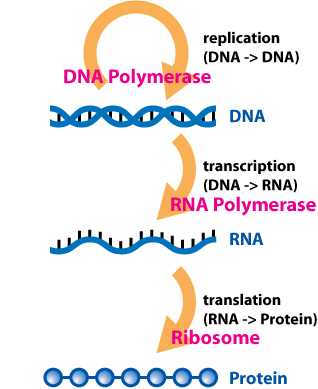
In biology, translation is the process in living cells in which proteins are produced using RNA molecules as templates. The generated protein is a sequence of amino acids. This sequence is determined by the sequence of nucleotides in the RNA. The nucleotides are considered three at a time. Each such triple results in addition of one specific amino acid to the protein being generated. The matching from nucleotide triple to amino acid is called the genetic code. The translation is performed by a large complex of functional RNA and proteins called ribosomes. The entire process is called gene expression.
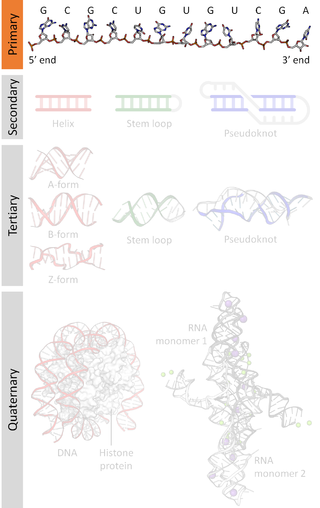
A nucleic acid sequence is a succession of bases within the nucleotides forming alleles within a DNA or RNA (GACU) molecule. This succession is denoted by a series of a set of five different letters that indicate the order of the nucleotides. By convention, sequences are usually presented from the 5' end to the 3' end. For DNA, with its double helix, there are two possible directions for the notated sequence; of these two, the sense strand is used. Because nucleic acids are normally linear (unbranched) polymers, specifying the sequence is equivalent to defining the covalent structure of the entire molecule. For this reason, the nucleic acid sequence is also termed the primary structure.

In molecular biology, a reading frame is a way of dividing the sequence of nucleotides in a nucleic acid molecule into a set of consecutive, non-overlapping triplets. Where these triplets equate to amino acids or stop signals during translation, they are called codons.
In computational biology, gene prediction or gene finding refers to the process of identifying the regions of genomic DNA that encode genes. This includes protein-coding genes as well as RNA genes, but may also include prediction of other functional elements such as regulatory regions. Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced.

A frameshift mutation is a genetic mutation caused by indels of a number of nucleotides in a DNA sequence that is not divisible by three. Due to the triplet nature of gene expression by codons, the insertion or deletion can change the reading frame, resulting in a completely different translation from the original. The earlier in the sequence the deletion or insertion occurs, the more altered the protein. A frameshift mutation is not the same as a single-nucleotide polymorphism in which a nucleotide is replaced, rather than inserted or deleted. A frameshift mutation will in general cause the reading of the codons after the mutation to code for different amino acids. The frameshift mutation will also alter the first stop codon encountered in the sequence. The polypeptide being created could be abnormally short or abnormally long, and will most likely not be functional.
The 5′ untranslated region is the region of a messenger RNA (mRNA) that is directly upstream from the initiation codon. This region is important for the regulation of translation of a transcript by differing mechanisms in viruses, prokaryotes and eukaryotes. While called untranslated, the 5′ UTR or a portion of it is sometimes translated into a protein product. This product can then regulate the translation of the main coding sequence of the mRNA. In many organisms, however, the 5′ UTR is completely untranslated, instead forming a complex secondary structure to regulate translation.

The start codon is the first codon of a messenger RNA (mRNA) transcript translated by a ribosome. The start codon always codes for methionine in eukaryotes and archaea and a N-formylmethionine (fMet) in bacteria, mitochondria and plastids.
In biology, the word gene has two meanings. The Mendelian gene is a basic unit of heredity. The molecular gene is a sequence of nucleotides in DNA, that is transcribed to produce a functional RNA. There are two types of molecular genes: protein-coding genes and non-coding genes.
In molecular biology and genetics, the sense of a nucleic acid molecule, particularly of a strand of DNA or RNA, refers to the nature of the roles of the strand and its complement in specifying a sequence of amino acids. Depending on the context, sense may have slightly different meanings. For example, the negative-sense strand of DNA is equivalent to the template strand, whereas the positive-sense strand is the non-template strand whose nucleotide sequence is equivalent to the sequence of the mRNA transcript.
In molecular genetics, an untranslated region refers to either of two sections, one on each side of a coding sequence on a strand of mRNA. If it is found on the 5' side, it is called the 5' UTR, or if it is found on the 3' side, it is called the 3' UTR. mRNA is RNA that carries information from DNA to the ribosome, the site of protein synthesis (translation) within a cell. The mRNA is initially transcribed from the corresponding DNA sequence and then translated into protein. However, several regions of the mRNA are usually not translated into protein, including the 5' and 3' UTRs.
Ribosomal frameshifting, also known as translational frameshifting or translational recoding, is a biological phenomenon that occurs during translation that results in the production of multiple, unique proteins from a single mRNA. The process can be programmed by the nucleotide sequence of the mRNA and is sometimes affected by the secondary, 3-dimensional mRNA structure. It has been described mainly in viruses, retrotransposons and bacterial insertion elements, and also in some cellular genes.

In molecular biology and genetics, DNA annotation or genome annotation is the process of describing the structure and function of the components of a genome, by analyzing and interpreting them in order to extract their biological significance and understand the biological processes in which they participate. Among other things, it identifies the locations of genes and all the coding regions in a genome and determines what those genes do.
The Consensus Coding Sequence (CCDS) Project is a collaborative effort to maintain a dataset of protein-coding regions that are identically annotated on the human and mouse reference genome assemblies. The CCDS project tracks identical protein annotations on the reference mouse and human genomes with a stable identifier, and ensures that they are consistently represented by the National Center for Biotechnology Information (NCBI), Ensembl, and UCSC Genome Browser. The integrity of the CCDS dataset is maintained through stringent quality assurance testing and on-going manual curation.
Periannan Senapathy is a molecular biologist, geneticist, author and entrepreneur. He is the founder, president and chief scientific officer at Genome International Corporation, a biotechnology, bioinformatics, and information technology firm based in Madison, Wisconsin, which develops computational genomics applications of next-generation DNA sequencing (NGS) and clinical decision support systems for analyzing patient genome data that aids in diagnosis and treatment of diseases.
An overlapping gene is a gene whose expressible nucleotide sequence partially overlaps with the expressible nucleotide sequence of another gene. In this way, a nucleotide sequence may make a contribution to the function of one or more gene products. Overlapping genes are present in and a fundamental feature of both cellular and viral genomes. The current definition of an overlapping gene varies significantly between eukaryotes, prokaryotes, and viruses. In prokaryotes and viruses overlap must be between coding sequences but not mRNA transcripts, and is defined when these coding sequences share a nucleotide on either the same or opposite strands. In eukaryotes, gene overlap is almost always defined as mRNA transcript overlap. Specifically, a gene overlap in eukaryotes is defined when at least one nucleotide is shared between the boundaries of the primary mRNA transcripts of two or more genes, such that a DNA base mutation at any point of the overlapping region would affect the transcripts of all genes involved. This definition includes 5′ and 3′ untranslated regions (UTRs) along with introns.
The vertebrate mitochondrial code is the genetic code found in the mitochondria of all vertebrata.
SEA-PHAGES stands for Science Education Alliance-Phage Hunters Advancing Genomics and Evolutionary Science; it was formerly called the National Genomics Research Initiative. This was the first initiative launched by the Howard Hughes Medical Institute (HHMI) Science Education Alliance (SEA) by their director Tuajuanda C. Jordan in 2008 to improve the retention of Science, technology, engineering, and mathematics (STEM) students. SEA-PHAGES is a two-semester undergraduate research program administered by the University of Pittsburgh's Graham Hatfull's group and the Howard Hughes Medical Institute's Science Education Division. Students from over 100 universities nationwide engage in authentic individual research that includes a wet-bench laboratory and a bioinformatics component.
The split gene theory is a theory of the origin of introns, long non-coding sequences in eukaryotic genes between the exons. The theory holds that the randomness of primordial DNA sequences would only permit small (< 600bp) open reading frames (ORFs), and that important intron structures and regulatory sequences are derived from stop codons. In this introns-first framework, the spliceosomal machinery and the nucleus evolved due to the necessity to join these ORFs into larger proteins, and that intronless bacterial genes are less ancestral than the split eukaryotic genes. The theory originated with Periannan Senapathy.

