History

Transcriptomics has been characterised by the development of new techniques which have redefined what is possible every decade or so and rendered previous technologies obsolete. The first attempt at capturing a partial human transcriptome was published in 1991 and reported 609 mRNA sequences from the human brain. [2] In 2008, two human transcriptomes, composed of millions of transcript-derived sequences covering 16,000 genes, were published, [3] [4] and by 2015 transcriptomes had been published for hundreds of individuals. [5] [6] Transcriptomes of different disease states, tissues, or even single cells are now routinely generated. [6] [7] [8] This explosion in transcriptomics has been driven by the rapid development of new technologies with improved sensitivity and economy. [9] [10] [11] [12]
Before transcriptomics
Studies of individual transcripts were being performed several decades before any transcriptomics approaches were available. Libraries of silkmoth mRNA transcripts were collected and converted to complementary DNA (cDNA) for storage using reverse transcriptase in the late 1970s. [13] In the 1980s, low-throughput sequencing using the Sanger method was used to sequence random transcripts, producing expressed sequence tags (ESTs). [2] [14] [15] [16] The Sanger method of sequencing was predominant until the advent of high-throughput methods such as sequencing by synthesis (Solexa/Illumina). ESTs came to prominence during the 1990s as an efficient method to determine the gene content of an organism without sequencing the entire genome. [16] Amounts of individual transcripts were quantified using Northern blotting, nylon membrane arrays, and later reverse transcriptase quantitative PCR (RT-qPCR) methods, [17] [18] but these methods are laborious and can only capture a tiny subsection of a transcriptome. [12] Consequently, the manner in which a transcriptome as a whole is expressed and regulated remained unknown until higher-throughput techniques were developed.
Early attempts
The word "transcriptome" was first used in the 1990s. [19] [20] In 1995, one of the earliest sequencing-based transcriptomic methods was developed, serial analysis of gene expression (SAGE), which worked by Sanger sequencing of concatenated random transcript fragments. [21] Transcripts were quantified by matching the fragments to known genes. A variant of SAGE using high-throughput sequencing techniques, called digital gene expression analysis, was also briefly used. [9] [22] However, these methods were largely overtaken by high throughput sequencing of entire transcripts, which provided additional information on transcript structure such as splice variants. [9]
Development of contemporary techniques
| RNA-Seq | Microarray | |
|---|---|---|
| Throughput | 1 day to 1 week per experiment [10] | 1–2 days per experiment [10] |
| Input RNA amount | Low ~ 1 ng total RNA [25] | High ~ 1 μg mRNA [26] |
| Labour intensity | High (sample preparation and data analysis) [10] [23] | Low [10] [23] |
| Prior knowledge | None required, although a reference genome/transcriptome sequence is useful [23] | Reference genome/transcriptome is required for design of probes [23] |
| Quantitation accuracy | ~90% (limited by sequence coverage) [27] | >90% (limited by fluorescence detection accuracy) [27] |
| Sequence resolution | RNA-Seq can detect SNPs and splice variants (limited by sequencing accuracy of ~99%) [27] | Specialised arrays can detect mRNA splice variants (limited by probe design and cross-hybridisation) [27] |
| Sensitivity | 1 transcript per million (approximate, limited by sequence coverage) [27] | 1 transcript per thousand (approximate, limited by fluorescence detection) [27] |
| Dynamic range | 100,000:1 (limited by sequence coverage) [28] | 1,000:1 (limited by fluorescence saturation) [28] |
| Technical reproducibility | >99% [29] [30] | >99% [31] [32] |
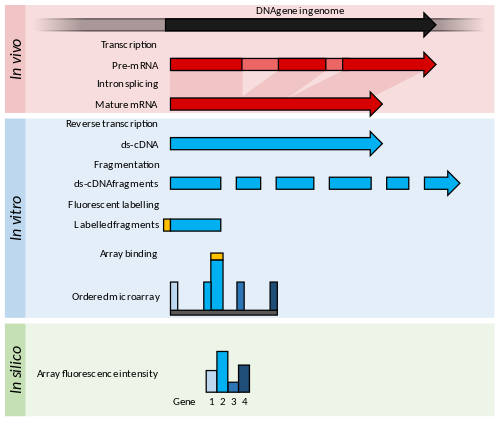
The dominant contemporary techniques, microarrays and RNA-Seq, were developed in the mid-1990s and 2000s. [9] [33] Microarrays that measure the abundances of a defined set of transcripts via their hybridisation to an array of complementary probes were first published in 1995. [34] [35] Microarray technology allowed the assay of thousands of transcripts simultaneously and at a greatly reduced cost per gene and labour saving. [36] Both spotted oligonucleotide arrays and Affymetrix high-density arrays were the method of choice for transcriptional profiling until the late 2000s. [12] [33] Over this period, a range of microarrays were produced to cover known genes in model or economically important organisms. Advances in design and manufacture of arrays improved the specificity of probes and allowed more genes to be tested on a single array. Advances in fluorescence detection increased the sensitivity and measurement accuracy for low abundance transcripts. [35] [37]
RNA-Seq is accomplished by reverse transcribing RNA in vitro and sequencing the resulting cDNAs. [10] Transcript abundance is derived from the number of counts from each transcript. The technique has therefore been heavily influenced by the development of high-throughput sequencing technologies. [9] [11] Massively parallel signature sequencing (MPSS) was an early example based on generating 16–20 bp sequences via a complex series of hybridisations, [38] [note 1] and was used in 2004 to validate the expression of ten thousand genes in Arabidopsis thaliana . [39] The earliest RNA-Seq work was published in 2006 with one hundred thousand transcripts sequenced using 454 technology. [40] This was sufficient coverage to quantify relative transcript abundance. RNA-Seq began to increase in popularity after 2008 when new Solexa/Illumina technologies allowed one billion transcript sequences to be recorded. [4] [10] [41] [42] This yield now allows for the quantification and comparison of human transcriptomes. [43]