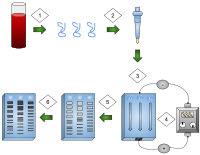
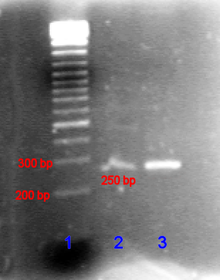
Agarose gel electrophoresis is a method of gel electrophoresis used in biochemistry, molecular biology, genetics, and clinical chemistry to separate a mixed population of macromolecules such as DNA or proteins in a matrix of agarose, one of the two main components of agar. The proteins may be separated by charge and/or size, and the DNA and RNA fragments by length. Biomolecules are separated by applying an electric field to move the charged molecules through an agarose matrix, and the biomolecules are separated by size in the agarose gel matrix.

Agarose is a heteropolysaccharide, generally extracted from certain red algae. It is a linear polymer made up of the repeating unit of agarobiose, which is a disaccharide made up of D-galactose and 3,6-anhydro-L-galactopyranose. Agarose is one of the two principal components of agar, and is purified from agar by removing agar's other component, agaropectin.
Molecular biology is a branch of biology that seeks to understand the molecular basis of biological activity in and between cells, including biomolecular synthesis, modification, mechanisms, and interactions.

Southern blot is a method used for detection and quantification of a specific DNA sequence in DNA samples. This method is used in molecular biology. Briefly, purified DNA from a biological sample is digested with restriction enzymes, and the resulting DNA fragments are separated by using an electric current to move them through a sieve-like gel or matrix, which allows smaller fragments to move faster than larger fragments. The DNA fragments are transferred out of the gel or matrix onto a solid membrane, which is then exposed to a DNA probe labeled with a radioactive, fluorescent, or chemical tag. The tag allows any DNA fragments containing complementary sequences with the DNA probe sequence to be visualized within the Southern blot.
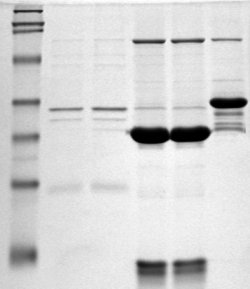
Polyacrylamide gel electrophoresis (PAGE) is a technique widely used in biochemistry, forensic chemistry, genetics, molecular biology and biotechnology to separate biological macromolecules, usually proteins or nucleic acids, according to their electrophoretic mobility. Electrophoretic mobility is a function of the length, conformation, and charge of the molecule. Polyacrylamide gel electrophoresis is a powerful tool used to analyze RNA samples. When polyacrylamide gel is denatured after electrophoresis, it provides information on the sample composition of the RNA species.

Gel electrophoresis of nucleic acids is an analytical technique to separate DNA or RNA fragments by size and reactivity. Nucleic acid molecules are placed on a gel, where an electric field induces the nucleic acids to migrate toward the positively charged anode. The molecules separate as they travel through the gel based on the each molecule's size and shape. Longer molecules move more slowly because they the gel resists their movement more forcefully than it resists shorter molecules. After some time, the electricity is turned off and the positions of the different molecules are analyzed.

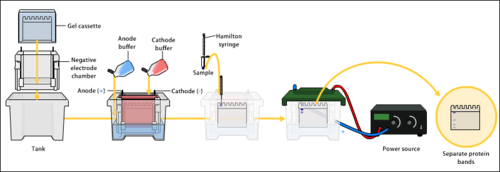
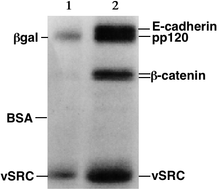
Protein electrophoresis is a method for analysing the proteins in a fluid or an extract. The electrophoresis may be performed with a small volume of sample in a number of alternative ways with or without a supporting medium, namely agarose or polyacrylamide. Variants of gel electrophoresis include SDS-PAGE, free-flow electrophoresis, electrofocusing, isotachophoresis, affinity electrophoresis, immunoelectrophoresis, counterelectrophoresis, and capillary electrophoresis. Each variant has many subtypes with individual advantages and limitations. Gel electrophoresis is often performed in combination with electroblotting or immunoblotting to give additional information about a specific protein.

In molecular biology and genetics, a blot is a method of transferring large biomolecules onto a carrier, such as a membrane composed of nitrocellulose, polyvinylidene fluoride or nylon. In many instances, this is done after a gel electrophoresis, transferring the molecules from the gel onto the blotting membrane, and other times adding the samples directly onto the membrane. After the blotting, the transferred molecules are then visualized by colorant staining, autoradiographic visualization of radiolabelled molecules, or specific labelling of some proteins or nucleic acids. The latter is done with antibodies or hybridization probes that bind only to some molecules of the blot and have an enzyme joined to them. After proper washing, this enzymatic activity is visualized by incubation with a proper reagent, rendering either a colored deposit on the blot or a chemiluminescent reaction which is registered by photographic film.

Temperature gradient gel electrophoresis (TGGE) and denaturing gradient gel electrophoresis (DGGE) are forms of electrophoresis which use either a temperature or chemical gradient to denature the sample as it moves across an acrylamide gel. TGGE and DGGE can be applied to nucleic acids such as DNA and RNA, and proteins. TGGE relies on temperature dependent changes in structure to separate nucleic acids. DGGE separates genes of the same size based on their different denaturing ability which is determined by their base pair sequence. DGGE was the original technique, and TGGE a refinement of it.
The southwestern blot, is a lab technique that involves identifying as well as characterizing DNA-binding proteins by their ability to bind to specific oligonucleotide probes. Determination of molecular weight of proteins binding to DNA is also made possible by the technique. The name originates from a combination of ideas underlying Southern blotting and Western blotting techniques of which they detect DNA and protein respectively. Similar to other types of blotting, proteins are separated by SDS-PAGE and are subsequently transferred to nitrocellulose membranes. Thereafter southwestern blotting begins to vary with regards to procedure as since the first blotting’s, many more have been proposed and discovered with goals of enhancing results. Former protocols were hampered by the need for large amounts of proteins and their susceptibility to degradation while being isolated.
Electrophoresis is the motion of charged dispersed particles or dissolved charged molecules relative to a fluid under the influence of a spatially uniform electric field.
TAE buffer is a buffer solution containing a mixture of Tris base, acetic acid and EDTA.

An electrophoretic mobility shift assay (EMSA) or mobility shift electrophoresis, also referred as a gel shift assay, gel mobility shift assay, band shift assay, or gel retardation assay, is a common affinity electrophoresis technique used to study protein–DNA or protein–RNA interactions. This procedure can determine if a protein or mixture of proteins is capable of binding to a given DNA or RNA sequence, and can sometimes indicate if more than one protein molecule is involved in the binding complex. Gel shift assays are often performed in vitro concurrently with DNase footprinting, primer extension, and promoter-probe experiments when studying transcription initiation, DNA gang replication, DNA repair or RNA processing and maturation, as well as pre-mRNA splicing. Although precursors can be found in earlier literature, most current assays are based on methods described by Garner and Revzin and Fried and Crothers.
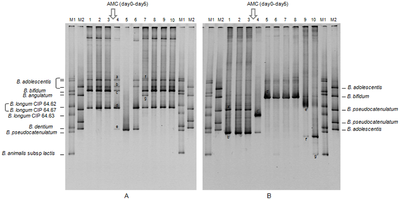
A molecular-weight size marker, also referred to as a protein ladder, DNA ladder, or RNA ladder, is a set of standards that are used to identify the approximate size of a molecule run on a gel during electrophoresis, using the principle that molecular weight is inversely proportional to migration rate through a gel matrix. Therefore, when used in gel electrophoresis, markers effectively provide a logarithmic scale by which to estimate the size of the other fragments.
QPNC-PAGE, or QuantitativePreparativeNativeContinuousPolyacrylamideGel Electrophoresis, is a bioanalytical, one-dimensional, high-resolution and high-precision electrophoresis technique applied in biochemistry and bioinorganic chemistry to separate proteins quantitatively by isoelectric point and by continuous elution from a gel column.

An electrophoretic color marker is a chemical used to monitor the progress of agarose gel electrophoresis and polyacrylamide gel electrophoresis (PAGE) since DNA, RNA, and most proteins are colourless. The color markers are made up of a mixture of dyes that migrate through the gel matrix alongside the sample of interest. They are typically designed to have different mobilities from the sample components and to generate colored bands that can be used to assess the migration and separation of sample components.

Affinity electrophoresis is a general name for many analytical methods used in biochemistry and biotechnology. Both qualitative and quantitative information may be obtained through affinity electrophoresis. Cross electrophoresis, the first affinity electrophoresis method, was created by Nakamura et al. Enzyme-substrate complexes have been detected using cross electrophoresis. The methods include the so-called electrophoretic mobility shift assay, charge shift electrophoresis and affinity capillary electrophoresis. The methods are based on changes in the electrophoretic pattern of molecules through biospecific interaction or complex formation. The interaction or binding of a molecule, charged or uncharged, will normally change the electrophoretic properties of a molecule. Membrane proteins may be identified by a shift in mobility induced by a charged detergent. Nucleic acids or nucleic acid fragments may be characterized by their affinity to other molecules. The methods have been used for estimation of binding constants, as for instance in lectin affinity electrophoresis or characterization of molecules with specific features like glycan content or ligand binding. For enzymes and other ligand-binding proteins, one-dimensional electrophoresis similar to counter electrophoresis or to "rocket immunoelectrophoresis", affinity electrophoresis may be used as an alternative quantification of the protein. Some of the methods are similar to affinity chromatography by use of immobilized ligands.
The northwestern blot, also known as the northwestern assay, is a hybrid analytical technique of the western blot and the northern blot, and is used in molecular biology to detect interactions between RNA and proteins. A related technique, the western blot, is used to detect a protein of interest that involves transferring proteins that are separated by gel electrophoresis onto a nitrocellulose membrane. A colored precipitate clusters along the band on the membrane containing a particular target protein. A northern blot is a similar analytical technique that, instead of detecting a protein of interest, is used to study gene expression by detection of RNA on a similar membrane. The northwestern blot combines the two techniques, and specifically involves the identification of labeled RNA that interact with proteins that are immobilized on a similar nitrocellulose membrane.

Discontinuous electrophoresis is a type of polyacrylamide gel electrophoresis. It was developed by Ornstein and Davis. This method produces high resolution and good band definition. It is widely used technique for separating proteins according to size and charge.
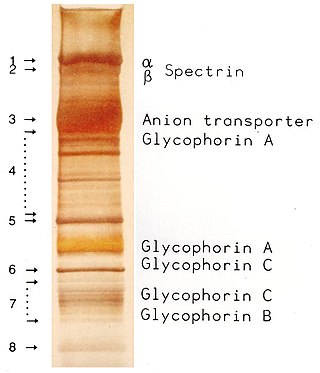
SDS-PAGE is a discontinuous electrophoretic system developed by Ulrich K. Laemmli which is commonly used as a method to separate proteins with molecular masses between 5 and 250 kDa. The combined use of sodium dodecyl sulfate and polyacrylamide gel eliminates the influence of structure and charge, and proteins are separated by differences in their size. At least up to 2012, the publication describing it was the most frequently cited paper by a single author, and the second most cited overall.