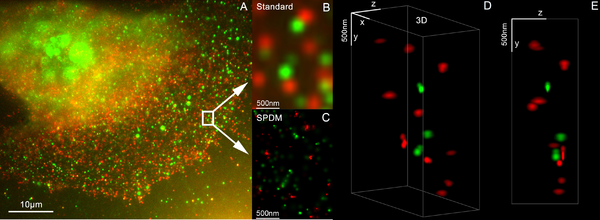
3D dual colour super resolution microscopy with Her2 and Her3 in breast cancer cells, standard dyes: Alexa 488, Alexa 568 - LIMON (SPDM +SMI
Vertico spatially modulated illumination (Vertico-SMI) is the fastest[citation needed]light microscope for the 3D analysis of complete cells in the nanometer range. It is based on two technologies developed in 1996, SMI (spatially modulated illumination) and SPDM (spectral precision distance microscopy). The effective optical resolution of this optical nanoscope has reached the vicinity of 5nm in 2D and 40nm in 3D, greatly surpassing the λ/2 resolution limit (about 200nm for blue light) applying to standard microscopy using transmission or reflection of natural light (as opposed to structured illumination or fluorescence) according to the Abbe resolution limit[1] That limit (also known as the Rayleigh limit) had been determined by Ernst Abbe in 1873 and governs the achievable resolution limit of microscopes using conventional techniques.
The Vertico-SMI microscope was developed by a team led by Christoph Cremer, emeritus[2] at Heidelberg University, and is based on the combination of light optical techniques of localization microscopy (SPDM, spectral precision distance microscopy) and structured illumination (SMI, spatially modulated illumination).
Since March 2008 many standard fluorescent dyes like GFP and Alexa fluorescent dyes can be used with this so-called SPDMphymod (physically modifiable fluorophores) localization microscopy, for which only one single laser wavelength of suitable intensity is sufficient for nanoimaging.
Configuration
SMI stands for a special type of laser optical illumination (spatially modulated illumination) and Vertico reflects the vertical arrangement of the microscope axis which renders possible the analysis of fixed cells but also of living cells with an optical resolution below 10 nanometers (1 nanometer = 1nm = 1 × 10−9m).
A particularity of this technology compared with focusing techniques such as 4Pi microscopy, is the wide field exposures which allow entire cells to be depicted at the nano scale. Such a 3D exposure of a whole cell with a typical object size of 20µm × 20µm require only 2 minutes. Wide field exposures signify that the entire object is illuminated and detected simultaneously.
SMI microscopy is a light optical process of the so-called point spread function-engineering. These are processes which modify the point spread function (PSF) of a microscope in a suitable manner to either increase the optical resolution, to maximize the precision of distance measurements of fluorescent objects that are small relative to the wavelength of the illuminating light, or to extract other structural parameters in the nanometer range.
The SMI microscope being developed at the Kirchhoff Institute for Physics at Heidelberg University achieves this in the following manner: The illumination intensity within the object range is not uniform, unlike conventional wide field fluorescence microscopes, but is spatially modulated in a precise manner by the use of two opposing interfering laser beams along the axis. The principle of the spatially modulated wave field was developed in 1993 by Bailey et al. The SMI microscopy approach used in the Heidelberg application moves the object in high-precision steps through the wave field, or the wave field itself is moved relative to the object by phase shift. This results in an improved axial size and distance resolution.[3][4]
SMI can be combined with other super resolution technologies, for instance with 3D LIMON or LSI-TIRF as a total internal reflection interferometer with laterally structured illumination. This SMI technique allowed to acquire light-optical images of autofluorophore distributions in the sections from human eye tissue with previously unmatched optical resolution. Use of three different excitation wavelengths (488, 568 and 647nm), enables to gather spectral information about the autofluorescence signal. This has been used for of human eye tissue affected by macular degeneration AMD.[5]
SPDM: localization microscopy
Single YFP molecule super resolution microscopy / SPDMphymod
A single, tiny source of light can be located much better than the resolution of a microscope: Although the light will produce a blurry spot, computer algorithms can be used to accurately calculate the center of the blurry spot, taking into account the point spread function of the microscope, the noise properties of the detector, and so on. However, this approach does not work when there are too many sources close to each other: The sources all blur together.
SPDM (spectral precision distance microscopy) is a family of techniques in fluorescence microscopy which gets around this problem by measuring just a few sources at a time, so that each source is "optically isolated" from the others (i.e., separated by more than the microscope's resolution, typically ~200-250nm).[6][7][8] Then, the above technique (finding the center of each blurry spot) can be used.
If the molecules have a variety of different spectra (absorption spectra and/or emission spectra), then it is possible to look at light from just a few molecules at a time by using the appropriate light sources and filters. Molecules can also be distinguished in more subtle ways based on fluorescent lifetime and other techniques.[6]
The structural resolution achievable using SPDM can be expressed in terms of the smallest measurable distance between two in their spatial position determined punctiform particle of different spectral characteristics ("topological resolution"). Modeling has shown that under suitable conditions regarding the precision of localization, particle density etc., the "topological resolution" corresponds to a "space frequency" which in terms of the classical definition is equivalent to a much improved optical resolution.
SPDM is a localization microscopy which achieves an effective optical resolution several times better than the conventional optical resolution (approx. 200-250nm), represented by the half-width of the main maximum of the effective point image function. By applying suitable laser optical precision processes, position and distances significantly smaller than the half-width of the point spread function (conventionally 200–250nm) can be measured with nanometer accuracy between targets with different spectral signatures.[6] An important area of application is genome research (study of the functional organization of the genome). Another important area of use is research into the structure of membranes.
One of the most important basics of the localization microscopy in general is the first experimental work for the localization of fluorescent objects in the nanoscale (3D) in 1996 [9] and theoretical and experimental proof for a localization accuracy using visible light in the range of 1nm – the basis for localization microscopy better than 1/100 of the wavelength.[10][11]
SPDMphymod: standard fluorescent dyes in the blinking mode like GFP
Dual color localization microscopy SPDMphymod/super resolution microscopy with GFP & RFP fusion proteins
Only in the past two years have molecules been used in nanoscopic studies which emit the same spectral light frequency (but with different spectral signatures based on the flashing characteristics) but which can be switched on and off by means of light as is necessary for spectral precision distance microscopy. By combining many thousands of images of the same cell, it was possible using laser optical precision measurements to record localization images with significantly improved optical resolution. The application of these novel nanoscopy processes appeared until recently very difficult because it was assumed that only specially manufactured molecules could be switched on and off in a suitable manner by using light.
In March 2008 Christoph Cremer’s lab discovered that this was also possible for many standard fluorescent dye like GFP, Alexa dyes and fluorescein molecules, provided certain photo-physical conditions are present. Using this so-called SPDMphymod (physically modifiable fluorophores) technology a single laser wavelength of suitable intensity is sufficient for nanoimaging. In contrast other localization microscopies need two laser wavelengths when special photo-switchable/photo-activatable fluorescence molecules are used.[12]
The GFP gene has been introduced and expressed in many procaryotic and eucaryotic cells and the Nobel Prize in Chemistry 2008 was awarded to Martin Chalfie, Osamu Shimomura, and Roger Y. Tsien for their discovery and development of the green fluorescent protein. The finding that these standard fluorescent molecules can be used extends the applicability of the SPMD method to numerous research fields in biophysics, cell biology and medicine.
Standard fluorescent dyes already successfully used with the SPDMphymod technology: GFP, RFP, YFP, Alexa 488, Alexa 568, Alexa 647, Cy2, Cy3, Atto 488 and fluorescein.
LIMON: 3D super resolution microscopy
LIMON (Light MicrOscopical nanosizing microscopy) was invented in 2001 at the University of Heidelberg and combines localization microscopy and spatially modulated illumination to the 3D super resolution microscopy.
The 3D images using Vertico-SMI are made possible by the combination of SMI and SPDM, whereby first the SMI and then the SPDM process is applied. The SMI process determines the center of particles and their spread in the direction of the microscope axis. While the center of particles/molecules can be determined with a 1–2nm precision, the spread around this point can be determined down to an axial diameter of approx. 30-40nm.
Subsequently, the lateral position of the individual particles/molecules is determined using SPDM, achieving a precision of a few nanometers. At present, SPDM achieves 16 frames/sec with an effective resolution of 10nm in 2D (object plane); approximately 2000 such frames are combined with SMI data (ca. 10 sec acquisition time) to achieve a three-dimensional image of highest resolution (effective optical 3D resolution ca. 40-50nm). With a faster camera, one can expect even higher rates (up to several hundred frames/sec, under development). Using suitable dyes, even higher effective optical 3D resolutions should be possible[13]
By combining SPDMphymod with SMI (both invented in Christoph Cremer´s lab in 1996) a 3D dual colour reconstruction of the spatial arrangements of Her2/neu and Her3 clusters was achieved. The positions in all three directions of the protein clusters could be determined with an accuracy of about 25nm.[14]
Use of super resolution microscopy in industry
Despite its use in biomedical labs, super resolution technologies could serve as important tools in pharmaceutical research. They could be especially helpful in the identification and valuation of targets. For example, biomolecular machines (BMM) are highly complex nanostructures consisting of several large molecules and which are responsible for basic functions in the body cells. Depending on their functional status, they have a defined 3D structure. Examples of biomolecular machines are nucleosomes which enable the DNA, a two meter long carrier of genetic information, to fold in the body cells in a space of a few millionth of a millimeter in diameter only. Therefore, the DNA can serve as an information and control center.
By using LIMON 3D in combination with LIMON complex labeling, it is possible for the first time to make hidden proteins or nucleic acids of a 3D-molecule complex of the so-called biomolecular machines visible without destroying the complex. Up to now, the problem in most cases was that the complex had to be destroyed for detailed analysis of the individual macromolecules therein. Alternatively, virtual computer simulation models or expensive nuclear magnetic resonance methods were used to visualize the three-dimensional structure of such complexes.[15]
↑ US patent 7,342,717: Christoph Cremer, Michael Hausmann, Joachim Bradl, Bernhard Schneider Wave field microscope with detection point spread function, priority date 10 July 1997
↑ Best G, Amberger R, Baddeley D, Ach T, Dithmar S, Heintzmann R, Cremer C (2011). "Structured illumination microscopy of autofluorescent aggregations in human tissue". Micron. 42 (4): 330–335. doi:10.1016/j.micron.2010.06.016. PMID20926302.{{cite journal}}: CS1 maint: multiple names: authors list (link)
↑ Bornfleth; Sätzler; Eils; Cremer (1 February 1998). "High-precision distance measurements and volume-conserving segmentation of objects near and below the resolution limit in three-dimensional confocal fluorescence microscopy". Journal of Microscopy. 189 (2): 118–136. doi:10.1046/j.1365-2818.1998.00276.x. S2CID73578516.
↑ Bradl J., Rinke B., Esa A., Edelmann P., Krieger H., Schneider B., Hausmann M., Cremer C. (1996). Bigio, Irving J; Grundfest, Warren S; Schneckenburger, Herbert; Svanberg, Katarina; Viallet, Pierre M (eds.). "Comparative study of three-dimensional localization accuracy in conventional, confocal laser scanning and axialtomographic fluorescence light microscopy". Proc. SPIE. Optical Biopsies and Microscopic Techniques. 2926: 201–206. Bibcode:1996SPIE.2926..201B. doi:10.1117/12.260797. S2CID55468495.{{cite journal}}: CS1 maint: multiple names: authors list (link)
↑ Heintzmann R., Münch H., Cremer C. (1997). "High-precision measurements in epifluorescent microscopy - simulation and experiment". Cell Vision. 4: 252–253.{{cite journal}}: CS1 maint: multiple names: authors list (link)
↑ US patent 6,424,421: Christoph Cremer, Michael Hausmann, Joachim Bradl, Bernd Rinke Method and devices for measuring distances between object structures, priority date 23 December 1996
↑ Manuel Gunkel, Fabian Erdel, Karsten Rippe, Paul Lemmer, Rainer Kaufmann, Christoph Hörmann, Roman Amberger and Christoph Cremer (2009): Dual color localization microscopy of cellular nanostructures. In: Biotechnology Journal, 2009, 4, 927–938. ISSN1860-6768
↑ Kaufmann Rainer, Müller Patrick, Hildenbrand Georg, Hausmann Michael, Cremer Christoph (2010). "Analysis of Her2/neu membrane protein clusters in different types of breast cancer cells using localization microscopy". Journal of Microscopy. 242 (1): 46–54. doi:10.1111/j.1365-2818.2010.03436.x. PMID21118230. S2CID2119158.{{cite journal}}: CS1 maint: multiple names: authors list (link)
Microscopy is the technical field of using microscopes to view objects and areas of objects that cannot be seen with the naked eye. There are three well-known branches of microscopy: optical, electron, and scanning probe microscopy, along with the emerging field of X-ray microscopy.
A microscope is a laboratory instrument used to examine objects that are too small to be seen by the naked eye. Microscopy is the science of investigating small objects and structures using a microscope. Microscopic means being invisible to the eye unless aided by a microscope.
The optical microscope, also referred to as a light microscope, is a type of microscope that commonly uses visible light and a system of lenses to generate magnified images of small objects. Optical microscopes are the oldest design of microscope and were possibly invented in their present compound form in the 17th century. Basic optical microscopes can be very simple, although many complex designs aim to improve resolution and sample contrast.
In optics, any optical instrument or system – a microscope, telescope, or camera – has a principal limit to its resolution due to the physics of diffraction. An optical instrument is said to be diffraction-limited if it has reached this limit of resolution performance. Other factors may affect an optical system's performance, such as lens imperfections or aberrations, but these are caused by errors in the manufacture or calculation of a lens, whereas the diffraction limit is the maximum resolution possible for a theoretically perfect, or ideal, optical system.
A total internal reflection fluorescence microscope (TIRFM) is a type of microscope with which a thin region of a specimen, usually less than 200 nanometers can be observed.
A fluorescence microscope is an optical microscope that uses fluorescence instead of, or in addition to, scattering, reflection, and attenuation or absorption, to study the properties of organic or inorganic substances. "Fluorescence microscope" refers to any microscope that uses fluorescence to generate an image, whether it is a simple set up like an epifluorescence microscope or a more complicated design such as a confocal microscope, which uses optical sectioning to get better resolution of the fluorescence image.
Confocal microscopy, most frequently confocal laser scanning microscopy (CLSM) or laser scanning confocal microscopy (LSCM), is an optical imaging technique for increasing optical resolution and contrast of a micrograph by means of using a spatial pinhole to block out-of-focus light in image formation. Capturing multiple two-dimensional images at different depths in a sample enables the reconstruction of three-dimensional structures within an object. This technique is used extensively in the scientific and industrial communities and typical applications are in life sciences, semiconductor inspection and materials science.
Two-photon excitation microscopy is a fluorescence imaging technique that is particularly well-suited to image scattering living tissue of up to about one millimeter in thickness. Unlike traditional fluorescence microscopy, where the excitation wavelength is shorter than the emission wavelength, two-photon excitation requires simultaneous excitation by two photons with longer wavelength than the emitted light. The laser is focused onto a specific location in the tissue and scanned across the sample to sequentially produce the image. Due to the non-linearity of two-photon excitation, mainly fluorophores in the micrometer-sized focus of the laser beam are excited, which results in the spatial resolution of the image. This contrasts with confocal microscopy, where the spatial resolution is produced by the interaction of excitation focus and the confined detection with a pinhole.
A 4Pi microscope is a laser scanning fluorescence microscope with an improved axial resolution. With it the typical range of the axial resolution of 500–700 nm can be improved to 100–150 nm, which corresponds to an almost spherical focal spot with 5–7 times less volume than that of standard confocal microscopy.
Near-field scanning optical microscopy (NSOM) or scanning near-field optical microscopy (SNOM) is a microscopy technique for nanostructure investigation that breaks the far field resolution limit by exploiting the properties of evanescent waves. In SNOM, the excitation laser light is focused through an aperture with a diameter smaller than the excitation wavelength, resulting in an evanescent field on the far side of the aperture. When the sample is scanned at a small distance below the aperture, the optical resolution of transmitted or reflected light is limited only by the diameter of the aperture. In particular, lateral resolution of 6 nm and vertical resolution of 2–5 nm have been demonstrated.
Nanophotonics or nano-optics is the study of the behavior of light on the nanometer scale, and of the interaction of nanometer-scale objects with light. It is a branch of optics, optical engineering, electrical engineering, and nanotechnology. It often involves dielectric structures such as nanoantennas, or metallic components, which can transport and focus light via surface plasmon polaritons.
RESOLFT, an acronym for REversible Saturable OpticaLFluorescence Transitions, denotes a group of optical fluorescence microscopy techniques with very high resolution. Using standard far field visible light optics a resolution far below the diffraction limit down to molecular scales can be obtained.
Christoph Cremer is a German physicist and emeritus at the Ruprecht-Karls-University Heidelberg, former honorary professor at the University of Mainz and was a former group leader at Institute of Molecular Biology (IMB) at the Johannes Gutenberg University of Mainz, Germany, who has successfully overcome the conventional limit of resolution that applies to light based investigations by a range of different methods. In the meantime, according to his own statement, Christoph Cremer is a member of the Max Planck Institute for Chemistry and the Max Planck Institute for Polymer Research.
Super-resolution microscopy is a series of techniques in optical microscopy that allow such images to have resolutions higher than those imposed by the diffraction limit, which is due to the diffraction of light. Super-resolution imaging techniques rely on the near-field or on the far-field. Among techniques that rely on the latter are those that improve the resolution only modestly beyond the diffraction-limit, such as confocal microscopy with closed pinhole or aided by computational methods such as deconvolution or detector-based pixel reassignment, the 4Pi microscope, and structured-illumination microscopy technologies such as SIM and SMI.
Multifocal plane microscopy (MUM), also known as multiplane microscopy or multifocus microscopy, is a form of light microscopy that allows the tracking of the 3D dynamics in live cells at high temporal and spatial resolution by simultaneously imaging different focal planes within the specimen. In this methodology, the light collected from the sample by an infinity-corrected objective lens is split into two paths. In each path the split light is focused onto a detector which is placed at a specific calibrated distance from the tube lens. In this way, each detector images a distinct plane within the sample. The first developed MUM setup was capable of imaging two distinct planes within the sample. However, the setup can be modified to image more than two planes by further splitting the light in each light path and focusing it onto detectors placed at specific calibrated distances. It has later been improved for imaging up to four distinct planes. To image a greater number of focal planes, simpler techniques based on image splitting optics have been developed. One example is by using a customized image splitting prism, which is capable of capturing up to 8 focal planes using only two cameras. Better yet, standard off-the-shelf partial beamsplitters can be used to construct a so-called z-splitter prism that allows simultaneous imaging of 9 individual focal planes using a single camera. Another technique called multifocus microscopy (MFM) uses diffractive Fourier optics to image up to 25 focal planes.
Photo-activated localization microscopy and stochastic optical reconstruction microscopy (STORM) are widefield fluorescence microscopy imaging methods that allow obtaining images with a resolution beyond the diffraction limit. The methods were proposed in 2006 in the wake of a general emergence of optical super-resolution microscopy methods, and were featured as Methods of the Year for 2008 by the Nature Methods journal. The development of PALM as a targeted biophysical imaging method was largely prompted by the discovery of new species and the engineering of mutants of fluorescent proteins displaying a controllable photochromism, such as photo-activatible GFP. However, the concomitant development of STORM, sharing the same fundamental principle, originally made use of paired cyanine dyes. One molecule of the pair, when excited near its absorption maximum, serves to reactivate the other molecule to the fluorescent state.
Light sheet fluorescence microscopy (LSFM) is a fluorescence microscopy technique with an intermediate-to-high optical resolution, but good optical sectioning capabilities and high speed. In contrast to epifluorescence microscopy only a thin slice of the sample is illuminated perpendicularly to the direction of observation. For illumination, a laser light-sheet is used, i.e. a laser beam which is focused only in one direction. A second method uses a circular beam scanned in one direction to create the lightsheet. As only the actually observed section is illuminated, this method reduces the photodamage and stress induced on a living sample. Also the good optical sectioning capability reduces the background signal and thus creates images with higher contrast, comparable to confocal microscopy. Because light sheet fluorescence microscopy scans samples by using a plane of light instead of a point, it can acquire images at speeds 100 to 1,000 times faster than those offered by point-scanning methods.
Lattice light-sheet microscopy is a modified version of light sheet fluorescence microscopy that increases image acquisition speed while decreasing damage to cells caused by phototoxicity. This is achieved by using a structured light sheet to excite fluorescence in successive planes of a specimen, generating a time series of 3D images which can provide information about dynamic biological processes.
Fluorescence imaging is a type of non-invasive imaging technique that can help visualize biological processes taking place in a living organism. Images can be produced from a variety of methods including: microscopy, imaging probes, and spectroscopy.
Structured illumination light sheet microscopy (SI-LSM) is an optical imaging technique used for achieving volumetric imaging with high temporal and spatial resolution in all three dimensions. It combines the ability of light sheet microscopy to maintain spatial resolution throughout relatively thick samples with the higher axial and spatial resolution characteristic of structured illumination microscopy. SI-LSM can achieve lateral resolution below 100 nm in biological samples hundreds of micrometers thick.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.