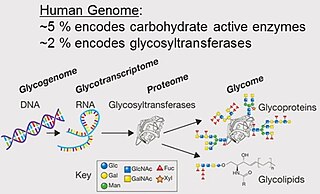
A glycome is the entire complement or complete set of all sugars, whether free or chemically bound in more complex molecules, of an organism. An alternative definition is the entirety of carbohydrates in a cell. The glycome may in fact be one of the most complex entities in nature. "Glycomics, analogous to genomics and proteomics, is the systematic study of all glycan structures of a given cell type or organism" and is a subset of glycobiology.

Mass spectrometry (MS) is an analytical technique that is used to measure the mass-to-charge ratio of ions. The results are presented as a mass spectrum, a plot of intensity as a function of the mass-to-charge ratio. Mass spectrometry is used in many different fields and is applied to pure samples as well as complex mixtures.

Tandem mass spectrometry, also known as MS/MS or MS2, is a technique in instrumental analysis where two or more stages of analysis using one or more mass analyzer are performed with an additional reaction step in between these analyses to increase their abilities to analyse chemical samples. A common use of tandem MS is the analysis of biomolecules, such as proteins and peptides.
Protein sequencing is the practical process of determining the amino acid sequence of all or part of a protein or peptide. This may serve to identify the protein or characterize its post-translational modifications. Typically, partial sequencing of a protein provides sufficient information to identify it with reference to databases of protein sequences derived from the conceptual translation of genes.

In mass spectrometry, matrix-assisted laser desorption/ionization (MALDI) is an ionization technique that uses a laser energy-absorbing matrix to create ions from large molecules with minimal fragmentation. It has been applied to the analysis of biomolecules and various organic molecules, which tend to be fragile and fragment when ionized by more conventional ionization methods. It is similar in character to electrospray ionization (ESI) in that both techniques are relatively soft ways of obtaining ions of large molecules in the gas phase, though MALDI typically produces far fewer multi-charged ions.
The terms glycans and polysaccharides are defined by IUPAC as synonyms meaning "compounds consisting of a large number of monosaccharides linked glycosidically". However, in practice the term glycan may also be used to refer to the carbohydrate portion of a glycoconjugate, such as a glycoprotein, glycolipid, or a proteoglycan, even if the carbohydrate is only an oligosaccharide. Glycans usually consist solely of O-glycosidic linkages of monosaccharides. For example, cellulose is a glycan composed of β-1,4-linked D-glucose, and chitin is a glycan composed of β-1,4-linked N-acetyl-D-glucosamine. Glycans can be homo- or heteropolymers of monosaccharide residues, and can be linear or branched.
Surface-enhanced laser desorption/ionization (SELDI) is a soft ionization method in mass spectrometry (MS) used for the analysis of protein mixtures. It is a variation of matrix-assisted laser desorption/ionization (MALDI). In MALDI, the sample is mixed with a matrix material and applied to a metal plate before irradiation by a laser, whereas in SELDI, proteins of interest in a sample become bound to a surface before MS analysis. The sample surface is a key component in the purification, desorption, and ionization of the sample. SELDI is typically used with time-of-flight (TOF) mass spectrometers and is used to detect proteins in tissue samples, blood, urine, or other clinical samples, however, SELDI technology can potentially be used in any application by simply modifying the sample surface.
Mascot is a software search engine that uses mass spectrometry data to identify proteins from peptide sequence databases. Mascot is widely used by research facilities around the world. Mascot uses a probabilistic scoring algorithm for protein identification that was adapted from the MOWSE algorithm. Mascot is freely available to use on the website of Matrix Science. A license is required for in-house use where more features can be incorporated.

Electron-transfer dissociation (ETD) is a method of fragmenting multiply-charged gaseous macromolecules in a mass spectrometer between the stages of tandem mass spectrometry (MS/MS). Similar to electron-capture dissociation, ETD induces fragmentation of large, multiply-charged cations by transferring electrons to them. ETD is used extensively with polymers and biological molecules such as proteins and peptides for sequence analysis. Transferring an electron causes peptide backbone cleavage into c- and z-ions while leaving labile post translational modifications (PTM) intact. The technique only works well for higher charge state peptide or polymer ions (z>2). However, relative to collision-induced dissociation (CID), ETD is advantageous for the fragmentation of longer peptides or even entire proteins. This makes the technique important for top-down proteomics. The method was developed by Hunt and coworkers at the University of Virginia.

MALDI mass spectrometry imaging (MALDI-MSI) is the use of matrix-assisted laser desorption ionization as a mass spectrometry imaging technique in which the sample, often a thin tissue section, is moved in two dimensions while the mass spectrum is recorded. Advantages, like measuring the distribution of a large amount of analytes at one time without destroying the sample, make it a useful method in tissue-based study.

Protein mass spectrometry refers to the application of mass spectrometry to the study of proteins. Mass spectrometry is an important method for the accurate mass determination and characterization of proteins, and a variety of methods and instrumentations have been developed for its many uses. Its applications include the identification of proteins and their post-translational modifications, the elucidation of protein complexes, their subunits and functional interactions, as well as the global measurement of proteins in proteomics. It can also be used to localize proteins to the various organelles, and determine the interactions between different proteins as well as with membrane lipids.

Time-of-flight mass spectrometry (TOFMS) is a method of mass spectrometry in which an ion's mass-to-charge ratio is determined by a time of flight measurement. Ions are accelerated by an electric field of known strength. This acceleration results in an ion having the same kinetic energy as any other ion that has the same charge. The velocity of the ion depends on the mass-to-charge ratio. The time that it subsequently takes for the ion to reach a detector at a known distance is measured. This time will depend on the velocity of the ion, and therefore is a measure of its mass-to-charge ratio. From this ratio and known experimental parameters, one can identify the ion.
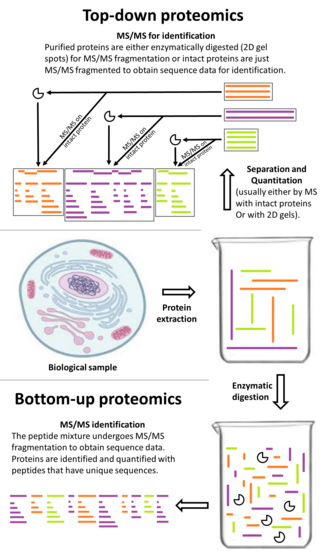
Top-down proteomics is a method of protein identification that either uses an ion trapping mass spectrometer to store an isolated protein ion for mass measurement and tandem mass spectrometry (MS/MS) analysis or other protein purification methods such as two-dimensional gel electrophoresis in conjunction with MS/MS. Top-down proteomics is capable of identifying and quantitating unique proteoforms through the analysis of intact proteins. The name is derived from the similar approach to DNA sequencing. During mass spectrometry intact proteins are typically ionized by electrospray ionization and trapped in a Fourier transform ion cyclotron resonance, quadrupole ion trap or Orbitrap mass spectrometer. Fragmentation for tandem mass spectrometry is accomplished by electron-capture dissociation or electron-transfer dissociation. Effective fractionation is critical for sample handling before mass-spectrometry-based proteomics. Proteome analysis routinely involves digesting intact proteins followed by inferred protein identification using mass spectrometry (MS). Top-down MS (non-gel) proteomics interrogates protein structure through measurement of an intact mass followed by direct ion dissociation in the gas phase.
Bottom-up proteomics is a common method to identify proteins and characterize their amino acid sequences and post-translational modifications by proteolytic digestion of proteins prior to analysis by mass spectrometry. The major alternative workflow used in proteomics is called top-down proteomics where intact proteins are purified prior to digestion and/or fragmentation either within the mass spectrometer or by 2D electrophoresis. Essentially, bottom-up proteomics is a relatively simple and reliable means of determining the protein make-up of a given sample of cells, tissues, etc.

Quantitative proteomics is an analytical chemistry technique for determining the amount of proteins in a sample. The methods for protein identification are identical to those used in general proteomics, but include quantification as an additional dimension. Rather than just providing lists of proteins identified in a certain sample, quantitative proteomics yields information about the physiological differences between two biological samples. For example, this approach can be used to compare samples from healthy and diseased patients. Quantitative proteomics is mainly performed by two-dimensional gel electrophoresis (2-DE), preparative native PAGE, or mass spectrometry (MS). However, a recent developed method of quantitative dot blot (QDB) analysis is able to measure both the absolute and relative quantity of an individual proteins in the sample in high throughput format, thus open a new direction for proteomic research. In contrast to 2-DE, which requires MS for the downstream protein identification, MS technology can identify and quantify the changes.
Sample preparation for mass spectrometry is used for the optimization of a sample for analysis in a mass spectrometer (MS). Each ionization method has certain factors that must be considered for that method to be successful, such as volume, concentration, sample phase, and composition of the analyte solution. Quite possibly the most important consideration in sample preparation is knowing what phase the sample must be in for analysis to be successful. In some cases the analyte itself must be purified before entering the ion source. In other situations, the matrix, or everything in the solution surrounding the analyte, is the most important factor to consider and adjust. Often, sample preparation itself for mass spectrometry can be avoided by coupling mass spectrometry to a chromatography method, or some other form of separation before entering the mass spectrometer. In some cases, the analyte itself must be adjusted so that analysis is possible, such as in protein mass spectrometry, where usually the protein of interest is cleaved into peptides before analysis, either by in-gel digestion or by proteolysis in solution.

Capillary electrophoresis–mass spectrometry (CE–MS) is an analytical chemistry technique formed by the combination of the liquid separation process of capillary electrophoresis with mass spectrometry. CE–MS combines advantages of both CE and MS to provide high separation efficiency and molecular mass information in a single analysis. It has high resolving power and sensitivity, requires minimal volume and can analyze at high speed. Ions are typically formed by electrospray ionization, but they can also be formed by matrix-assisted laser desorption/ionization or other ionization techniques. It has applications in basic research in proteomics and quantitative analysis of biomolecules as well as in clinical medicine. Since its introduction in 1987, new developments and applications have made CE-MS a powerful separation and identification technique. Use of CE–MS has increased for protein and peptides analysis and other biomolecules. However, the development of online CE–MS is not without challenges. Understanding of CE, the interface setup, ionization technique and mass detection system is important to tackle problems while coupling capillary electrophoresis to mass spectrometry.
In bio-informatics, a peptide-mass fingerprint or peptide-mass map is a mass spectrum of a mixture of peptides that comes from a digested protein being analyzed. The mass spectrum serves as a fingerprint in the sense that it is a pattern that can serve to identify the protein. The method for forming a peptide-mass fingerprint, developed in 1993, consists of isolating a protein, breaking it down into individual peptides, and determining the masses of the peptides through some form of mass spectrometry. Once formed, a peptide-mass fingerprint can be used to search in databases for related protein or even genomic sequences, making it a powerful tool for annotation of protein-coding genes.
Electrostatic spray ionization (ESTASI) is an ambient ionization method for mass spectrometry (MS) analysis of samples located on a flat or porous surface, or inside a microchannel. It was developed in 2011 by Professor Hubert H. Girault’s group at the École Polytechnique Fédérale de Lausanne (EPFL) in Switzerland. In a typical ESTASI process, a droplet of a protic solvent containing analytes is deposited on a sample area of interest which itself is mounted to an insulating substrate. Under this substrate and right below the droplet, an electrode is placed and connected with a pulsed high voltage (HV) to electrostatically charge the droplet during pulsing. When the electrostatic pressure is larger than the surface tension, droplets and ions are sprayed. ESTASI is a contactless process based on capacitive coupling. One advantage of ESTASI is, that the electrode and sample droplet act contact-less avoiding thereby any oxidation or reduction of the sample compounds at the electrode surface, which often happens during standard electrospray ionization (ESI). ESTASI is a powerful new ambient ionization technique that has already found many applications in the detection of different analytes, such as organic molecules, peptides and proteins with molecule weight up to 70 kDa. Furthermore, it was used to couple MS with various separation techniques including capillary electrophoresis and gel isoelectric focusing, and it was successfully applied under atmospheric pressure to the direct analysis of samples with only few preparation steps.
Zooarchaeology by mass spectrometry, commonly referred to by the abbreviation ZooMS, is a scientific method that identifies animal species by means of characteristic peptide sequences in the protein collagen. ZooMS is the most common archaeological application of peptide mass fingerprinting (PMF) and can be used for species identification of bones, teeth, skin and antler. It is commonly used to identify objects that cannot be identified morphologically. In an archaeological context this usually means that the object is too fragmented or that it has been shaped into an artefact. Archaeologists use these species identification to study among others past environments, diet and raw material selection for the production of tools.
