In computational biology, gene prediction or gene finding refers to the process of identifying the regions of genomic DNA that encode genes. This includes protein-coding genes as well as RNA genes, but may also include prediction of other functional elements such as regulatory regions. Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced.
In its earliest days, "gene finding" was based on painstaking experimentation on living cells and organisms. Statistical analysis of the rates of homologous recombination of several different genes could determine their order on a certain chromosome, and information from many such experiments could be combined to create a genetic map specifying the rough location of known genes relative to each other. Today, with comprehensive genome sequence and powerful computational resources at the disposal of the research community, gene finding has been redefined as a largely computational problem.
Determining that a sequence is functional should be distinguished from determining the function of the gene or its product. Predicting the function of a gene and confirming that the gene prediction is accurate still demands in vivo experimentation[1] through gene knockout and other assays, although frontiers of bioinformatics research [2] are making it increasingly possible to predict the function of a gene based on its sequence alone.
Gene prediction is one of the key steps in genome annotation, following sequence assembly, the filtering of non-coding regions and repeat masking.[3]
In empirical (similarity, homology or evidence-based) gene finding systems, the target genome is searched for sequences that are similar to extrinsic evidence in the form of the known expressed sequence tags, messenger RNA (mRNA), protein products, and homologous or orthologous sequences. Given an mRNA sequence, it is trivial to derive a unique genomic DNA sequence from which it had to have been transcribed. Given a protein sequence, a family of possible coding DNA sequences can be derived by reverse translation of the genetic code. Once candidate DNA sequences have been determined, it is a relatively straightforward algorithmic problem to efficiently search a target genome for matches, complete or partial, and exact or inexact. Given a sequence, local alignment algorithms such as BLAST, FASTA and Smith-Waterman look for regions of similarity between the target sequence and possible candidate matches. Matches can be complete or partial, and exact or inexact. The success of this approach is limited by the contents and accuracy of the sequence database.
A high degree of similarity to a known messenger RNA or protein product is strong evidence that a region of a target genome is a protein-coding gene. However, to apply this approach systemically requires extensive sequencing of mRNA and protein products. Not only is this expensive, but in complex organisms, only a subset of all genes in the organism's genome are expressed at any given time, meaning that extrinsic evidence for many genes is not readily accessible in any single cell culture. Thus, to collect extrinsic evidence for most or all of the genes in a complex organism requires the study of many hundreds or thousands of cell types, which presents further difficulties. For example, some human genes may be expressed only during development as an embryo or fetus, which might be difficult to study for ethical reasons.
Despite these difficulties, extensive transcript and protein sequence databases have been generated for human as well as other important model organisms in biology, such as mice and yeast. For example, the RefSeq database contains transcript and protein sequence from many different species, and the Ensembl system comprehensively maps this evidence to human and several other genomes. It is, however, likely that these databases are both incomplete and contain small but significant amounts of erroneous data.
New high-throughput transcriptome sequencing technologies such as RNA-Seq and ChIP-sequencing open opportunities for incorporating additional extrinsic evidence into gene prediction and validation, and allow structurally rich and more accurate alternative to previous methods of measuring gene expression such as expressed sequence tag or DNA microarray.
Major challenges involved in gene prediction involve dealing with sequencing errors in raw DNA data, dependence on the quality of the sequence assembly, handling short reads, frameshift mutations, overlapping genes and incomplete genes.
In prokaryotes it's essential to consider horizontal gene transfer when searching for gene sequence homology. An additional important factor underused in current gene detection tools is existence of gene clusters — operons (which are functioning units of DNA containing a cluster of genes under the control of a single promoter) in both prokaryotes and eukaryotes. Most popular gene detectors treat each gene in isolation, independent of others, which is not biologically accurate.
Ab initio methods
Ab Initio gene prediction is an intrinsic method based on gene content and signal detection. Because of the inherent expense and difficulty in obtaining extrinsic evidence for many genes, it is also necessary to resort to ab initio gene finding, in which the genomicDNA sequence alone is systematically searched for certain tell-tale signs of protein-coding genes. These signs can be broadly categorized as either signals, specific sequences that indicate the presence of a gene nearby, or content, statistical properties of the protein-coding sequence itself. Ab initio gene finding might be more accurately characterized as gene prediction, since extrinsic evidence is generally required to conclusively establish that a putative gene is functional.
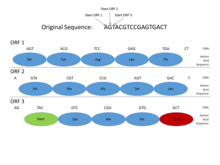
This picture shows how Open Reading Frames (ORFs) can be used for gene prediction. Gene prediction is the process of determining where a coding gene might be in a genomic sequence. Functional proteins must begin with a Start codon (where DNA transcription begins), and end with a Stop codon (where transcription ends). By looking at where those codons might fall in a DNA sequence, one can see where a functional protein might be located. This is important in gene prediction because it can reveal where coding genes are in an entire genomic sequence. In this example, a functional protein can be discovered using ORF3 because it begins with a Start codon, has multiple amino acids, and then ends with a Stop codon, all within the same reading frame.
In the genomes of prokaryotes, genes have specific and relatively well-understood promoter sequences (signals), such as the Pribnow box and transcription factorbinding sites, which are easy to systematically identify. Also, the sequence coding for a protein occurs as one contiguous open reading frame (ORF), which is typically many hundred or thousands of base pairs long. The statistics of stop codons are such that even finding an open reading frame of this length is a fairly informative sign. (Since 3 of the 64 possible codons in the genetic code are stop codons, one would expect a stop codon approximately every 20–25 codons, or 60–75 base pairs, in a random sequence.) Furthermore, protein-coding DNA has certain periodicities and other statistical properties that are easy to detect in a sequence of this length. These characteristics make prokaryotic gene finding relatively straightforward, and well-designed systems are able to achieve high levels of accuracy.
Ab initio gene finding in eukaryotes, especially complex organisms like humans, is considerably more challenging for several reasons. First, the promoter and other regulatory signals in these genomes are more complex and less well-understood than in prokaryotes, making them more difficult to reliably recognize. Two classic examples of signals identified by eukaryotic gene finders are CpG islands and binding sites for a poly(A) tail.
Second, splicing mechanisms employed by eukaryotic cells mean that a particular protein-coding sequence in the genome is divided into several parts (exons), separated by non-coding sequences (introns). (Splice sites are themselves another signal that eukaryotic gene finders are often designed to identify.) A typical protein-coding gene in humans might be divided into a dozen exons, each less than two hundred base pairs in length, and some as short as twenty to thirty. It is therefore much more difficult to detect periodicities and other known content properties of protein-coding DNA in eukaryotes.
Advanced gene finders for both prokaryotic and eukaryotic genomes typically use complex probabilistic models, such as hidden Markov models (HMMs) to combine information from a variety of different signal and content measurements. The GLIMMER system is a widely used and highly accurate gene finder for prokaryotes. GeneMark is another popular approach. Eukaryotic ab initio gene finders, by comparison, have achieved only limited success; notable examples are the GENSCAN and geneid programs. The SNAP gene finder is HMM-based like Genscan, and attempts to be more adaptable to different organisms, addressing problems related to using a gene finder on a genome sequence that it was not trained against.[7] A few recent approaches like mSplicer,[8] CONTRAST,[9] or mGene[10] also use machine learning techniques like support vector machines for successful gene prediction. They build a discriminative model using hidden Markov support vector machines or conditional random fields to learn an accurate gene prediction scoring function.
Ab Initio methods have been benchmarked, with some approaching 100% sensitivity,[3] however as the sensitivity increases, accuracy suffers as a result of increased false positives.
It has been suggested that signals other than those directly detectable in sequences may improve gene prediction. For example, the role of secondary structure in the identification of regulatory motifs has been reported.[12] In addition, it has been suggested that RNA secondary structure prediction helps splice site prediction.[13][14][15][16]
Neural networks
Artificial neural networks are computational models that excel at machine learning and pattern recognition. Neural networks must be trained with example data before being able to generalise for experimental data, and tested against benchmark data. Neural networks are able to come up with approximate solutions to problems that are hard to solve algorithmically, provided there is sufficient training data. When applied to gene prediction, neural networks can be used alongside other ab initio methods to predict or identify biological features such as splice sites.[17] One approach[18] involves using a sliding window, which traverses the sequence data in an overlapping manner. The output at each position is a score based on whether the network thinks the window contains a donor splice site or an acceptor splice site. Larger windows offer more accuracy but also require more computational power. A neural network is an example of a signal sensor as its goal is to identify a functional site in the genome.
Combined approaches
Programs such as Maker combine extrinsic and ab initio approaches by mapping protein and EST data to the genome to validate ab initio predictions. Augustus, which may be used as part of the Maker pipeline, can also incorporate hints in the form of EST alignments or protein profiles to increase the accuracy of the gene prediction.
Comparative genomics approaches
As the entire genomes of many different species are sequenced, a promising direction in current research on gene finding is a comparative genomics approach.
This is based on the principle that the forces of natural selection cause genes and other functional elements to undergo mutation at a slower rate than the rest of the genome, since mutations in functional elements are more likely to negatively impact the organism than mutations elsewhere. Genes can thus be detected by comparing the genomes of related species to detect this evolutionary pressure for conservation. This approach was first applied to the mouse and human genomes, using programs such as SLAM, SGP and TWINSCAN/N-SCAN and CONTRAST.[19]
Multiple informants
TWINSCAN examined only human-mouse synteny to look for orthologous genes. Programs such as N-SCAN and CONTRAST allowed the incorporation of alignments from multiple organisms, or in the case of N-SCAN, a single alternate organism from the target. The use of multiple informants can lead to significant improvements in accuracy.[19]
CONTRAST is composed of two elements. The first is a smaller classifier, identifying donor splice sites and acceptor splice sites as well as start and stop codons. The second element involves constructing a full model using machine learning. Breaking the problem into two means that smaller targeted data sets can be used to train the classifiers, and that classifier can operate independently and be trained with smaller windows. The full model can use the independent classifier, and not have to waste computational time or model complexity re-classifying intron-exon boundaries. The paper in which CONTRAST is introduced proposes that their method (and those of TWINSCAN, etc.) be classified as de novo gene assembly, using alternate genomes, and identifying it as distinct from ab initio, which uses a target 'informant' genomes.[19]
Comparative gene finding can also be used to project high quality annotations from one genome to another. Notable examples include Projector, GeneWise, GeneMapper and GeMoMa. Such techniques now play a central role in the annotation of all genomes.
Pseudogene prediction
Pseudogenes are close relatives of genes, sharing very high sequence homology, but being unable to code for the same protein product. Whilst once relegated as byproducts of gene sequencing, increasingly, as regulatory roles are being uncovered, they are becoming predictive targets in their own right.[20] Pseudogene prediction utilises existing sequence similarity and ab initio methods, whilst adding additional filtering and methods of identifying pseudogene characteristics.
Sequence similarity methods can be customised for pseudogene prediction using additional filtering to find candidate pseudogenes. This could use disablement detection, which looks for nonsense or frameshift mutations that would truncate or collapse an otherwise functional coding sequence.[21] Additionally, translating DNA into proteins sequences can be more effective than just straight DNA homology.[20]
Content sensors can be filtered according to the differences in statistical properties between pseudogenes and genes, such as a reduced count of CpG islands in pseudogenes, or the differences in G-C content between pseudogenes and their neighbours. Signal sensors also can be honed to pseudogenes, looking for the absence of introns or polyadenine tails. [22]
Metagenomic gene prediction
Metagenomics is the study of genetic material recovered from the environment, resulting in sequence information from a pool of organisms. Predicting genes is useful for comparative metagenomics.
Metagenomics tools also fall into the basic categories of using either sequence similarity approaches (MEGAN4) and ab initio techniques (GLIMMER-MG).
Glimmer-MG[23] is an extension to GLIMMER that relies mostly on an ab initio approach for gene finding and by using training sets from related organisms. The prediction strategy is augmented by classification and clustering gene data sets prior to applying ab initio gene prediction methods. The data is clustered by species. This classification method leverages techniques from metagenomic phylogenetic classification. An example of software for this purpose is, Phymm, which uses interpolated markov models—and PhymmBL, which integrates BLAST into the classification routines.
MEGAN4[24] uses a sequence similarity approach, using local alignment against databases of known sequences, but also attempts to classify using additional information on functional roles, biological pathways and enzymes. As in single organism gene prediction, sequence similarity approaches are limited by the size of the database.
FragGeneScan and MetaGeneAnnotator are popular gene prediction programs based on Hidden Markov model. These predictors account for sequencing errors, partial genes and work for short reads.
Another fast and accurate tool for gene prediction in metagenomes is MetaGeneMark.[25] This tool is used by the DOE Joint Genome Institute to annotate IMG/M, the largest metagenome collection to date.
Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.
The human genome is a complete set of nucleic acid sequences for humans, encoded as DNA within the 23 chromosome pairs in cell nuclei and in a small DNA molecule found within individual mitochondria. These are usually treated separately as the nuclear genome and the mitochondrial genome. Human genomes include both protein-coding DNA sequences and various types of DNA that does not encode proteins. The latter is a diverse category that includes DNA coding for non-translated RNA, such as that for ribosomal RNA, transfer RNA, ribozymes, small nuclear RNAs, and several types of regulatory RNAs. It also includes promoters and their associated gene-regulatory elements, DNA playing structural and replicatory roles, such as scaffolding regions, telomeres, centromeres, and origins of replication, plus large numbers of transposable elements, inserted viral DNA, non-functional pseudogenes and simple, highly repetitive sequences. Introns make up a large percentage of non-coding DNA. Some of this non-coding DNA is non-functional junk DNA, such as pseudogenes, but there is no firm consensus on the total amount of junk DNA.
The coding region of a gene, also known as the coding sequence(CDS), is the portion of a gene's DNA or RNA that codes for protein. Studying the length, composition, regulation, splicing, structures, and functions of coding regions compared to non-coding regions over different species and time periods can provide a significant amount of important information regarding gene organization and evolution of prokaryotes and eukaryotes. This can further assist in mapping the human genome and developing gene therapy.
Alternative splicing, or alternative RNA splicing, or differential splicing, is an alternative splicing process during gene expression that allows a single gene to code for multiple proteins. In this process, particular exons of a gene may be included within or excluded from the final, processed messenger RNA (mRNA) produced from that gene. This means the exons are joined in different combinations, leading to different (alternative) mRNA strands. Consequently, the proteins translated from alternatively spliced mRNAs usually contain differences in their amino acid sequence and, often, in their biological functions.
Codon usage bias refers to differences in the frequency of occurrence of synonymous codons in coding DNA. A codon is a series of three nucleotides that encodes a specific amino acid residue in a polypeptide chain or for the termination of translation.
Pseudogenes are nonfunctional segments of DNA that resemble functional genes. Most arise as superfluous copies of functional genes, either directly by gene duplication or indirectly by reverse transcription of an mRNA transcript. Pseudogenes are usually identified when genome sequence analysis finds gene-like sequences that lack regulatory sequences needed for transcription or translation, or whose coding sequences are obviously defective due to frameshifts or premature stop codons. Pseudogenes are a type of junk DNA.
In bioinformatics, sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of a wide range of analytical methods to understand its features, function, structure, or evolution. Methodologies used include sequence alignment, searches against biological databases, and others.
In molecular biology, open reading frames (ORFs) are defined as spans of DNA sequence between the start and stop codons. Usually, this is considered within a studied region of a prokaryotic DNA sequence, where only one of the six possible reading frames will be "open". Such an ORF may contain a start codon and by definition cannot extend beyond a stop codon. That start codon indicates where translation may start. The transcription termination site is located after the ORF, beyond the translation stop codon. If transcription were to cease before the stop codon, an incomplete protein would be made during translation.
Metagenomics is the study of genetic material recovered directly from environmental or clinical samples by a method called sequencing. The broad field may also be referred to as environmental genomics, ecogenomics, community genomics or microbiomics.
In biology, the word gene can have several different meanings. The Mendelian gene is a basic unit of heredity and the molecular gene is a sequence of nucleotides in DNA that is transcribed to produce a functional RNA. There are two types of molecular genes: protein-coding genes and non-coding genes.
GeneMark is a generic name for a family of ab initio gene prediction algorithms and software programs developed at the Georgia Institute of Technology in Atlanta. Developed in 1993, original GeneMark was used in 1995 as a primary gene prediction tool for annotation of the first completely sequenced bacterial genome of Haemophilus influenzae, and in 1996 for the first archaeal genome of Methanococcus jannaschii. The algorithm introduced inhomogeneous three-periodic Markov chain models of protein-coding DNA sequence that became standard in gene prediction as well as Bayesian approach to gene prediction in two DNA strands simultaneously. Species specific parameters of the models were estimated from training sets of sequences of known type. The major step of the algorithm computes for a given DNA fragment posterior probabilities of either being "protein-coding" in each of six possible reading frames or being "non-coding". The original GeneMark was an HMM-like algorithm; it could be viewed as approximation to known in the HMM theory posterior decoding algorithm for appropriately defined HMM model of DNA sequence.
GENCODE is a scientific project in genome research and part of the ENCODE scale-up project.
A nested gene is a gene whose entire coding sequence lies within the bounds of a larger external gene. The coding sequence for a nested gene differs greatly from the coding sequence for its external host gene. Typically, nested genes and their host genes encode functionally unrelated proteins, and have different expression patterns in an organism.
In molecular biology and genetics, DNA annotation or genome annotation is the process of describing the structure and function of the components of a genome, by analyzing and interpreting them in order to extract their biological significance and understand the biological processes in which they participate. Among other things, it identifies the locations of genes and all the coding regions in a genome and determines what those genes do.
Periannan Senapathy is a molecular biologist, geneticist, author and entrepreneur. He is the founder, president and chief scientific officer at Genome International Corporation, a biotechnology, bioinformatics, and information technology firm based in Madison, Wisconsin, which develops computational genomics applications of next-generation DNA sequencing (NGS) and clinical decision support systems for analyzing patient genome data that aids in diagnosis and treatment of diseases.
De novo transcriptome assembly is the de novo sequence assembly method of creating a transcriptome without the aid of a reference genome.
WormBase is an online biological database about the biology and genome of the nematode model organism Caenorhabditis elegans and contains information about other related nematodes. WormBase is used by the C. elegans research community both as an information resource and as a place to publish and distribute their results. The database is regularly updated with new versions being released every two months. WormBase is one of the organizations participating in the Generic Model Organism Database (GMOD) project.
Machine learning in bioinformatics is the application of machine learning algorithms to bioinformatics, including genomics, proteomics, microarrays, systems biology, evolution, and text mining.
The split gene theory is a theory of the origin of introns, long non-coding sequences in eukaryotic genes between the exons. The theory holds that the randomness of primordial DNA sequences would only permit small (< 600bp) open reading frames (ORFs), and that important intron structures and regulatory sequences are derived from stop codons. In this introns-first framework, the spliceosomal machinery and the nucleus evolved due to the necessity to join these ORFs into larger proteins, and that intronless bacterial genes are less ancestral than the split eukaryotic genes. The theory originated with Periannan Senapathy.
References
↑ Sleator RD (August 2010). "An overview of the current status of eukaryote gene prediction strategies". Gene. 461 (1–2): 1–4. doi:10.1016/j.gene.2010.04.008. PMID20430068.
↑ Patterson DJ, Yasuhara K, Ruzzo WL (2002). "Pre-mRNA secondary structure prediction aids splice site prediction". Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing: 223–34. PMID11928478.
↑ Marashi SA, Goodarzi H, Sadeghi M, Eslahchi C, Pezeshk H (February 2006). "Importance of RNA secondary structure information for yeast donor and acceptor splice site predictions by neural networks". Computational Biology and Chemistry. 30 (1): 50–7. doi:10.1016/j.compbiolchem.2005.10.009. PMID16386465.
↑ Zhang Z, Gerstein M (August 2004). "Large-scale analysis of pseudogenes in the human genome". Current Opinion in Genetics & Development. 14 (4): 328–35. doi:10.1016/j.gde.2004.06.003. PMID15261647.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.