Related Research Articles

In biology, epigenetics is the study of heritable traits, or a stable change of cell function, that happen without changes to the DNA sequence. The Greek prefix epi- in epigenetics implies features that are "on top of" or "in addition to" the traditional genetic mechanism of inheritance. Epigenetics usually involves a change that is not erased by cell division, and affects the regulation of gene expression. Such effects on cellular and physiological phenotypic traits may result from environmental factors, or be part of normal development. They can lead to cancer.

DNA repair is a collection of processes by which a cell identifies and corrects damage to the DNA molecules that encode its genome. In human cells, both normal metabolic activities and environmental factors such as radiation can cause DNA damage, resulting in tens of thousands of individual molecular lesions per cell per day. Many of these lesions cause structural damage to the DNA molecule and can alter or eliminate the cell's ability to transcribe the gene that the affected DNA encodes. Other lesions induce potentially harmful mutations in the cell's genome, which affect the survival of its daughter cells after it undergoes mitosis. As a consequence, the DNA repair process is constantly active as it responds to damage in the DNA structure. When normal repair processes fail, and when cellular apoptosis does not occur, irreparable DNA damage may occur. This can eventually lead to malignant tumors, or cancer as per the two-hit hypothesis.

A molecular lesion or point lesion is damage to the structure of a biological molecule such as DNA, RNA, or protein. This damage may result in the reduction or absence of normal function, and in rare cases the gain of a new function. Lesions in DNA may consist of breaks or other changes in chemical structure of the helix, ultimately preventing transcription. Meanwhile, lesions in proteins consist of both broken bonds and improper folding of the amino acid chain. While many nucleic acid lesions are general across DNA and RNA, some are specific to one, such as thymine dimers being found exclusively in DNA. Several cellular repair mechanisms exist, ranging from global to specific, in order to prevent lasting damage resulting from lesions.

A germline mutation, or germinal mutation, is any detectable variation within germ cells. Mutations in these cells are the only mutations that can be passed on to offspring, when either a mutated sperm or oocyte come together to form a zygote. After this fertilization event occurs, germ cells divide rapidly to produce all of the cells in the body, causing this mutation to be present in every somatic and germline cell in the offspring; this is also known as a constitutional mutation. Germline mutation is distinct from somatic mutation.

A neoplasm is a type of abnormal and excessive growth of tissue. The process that occurs to form or produce a neoplasm is called neoplasia. The growth of a neoplasm is uncoordinated with that of the normal surrounding tissue, and persists in growing abnormally, even if the original trigger is removed. This abnormal growth usually forms a mass, which may be called a tumour or tumor.

Triple-stranded DNA is a DNA structure in which three oligonucleotides wind around each other and form a triple helix. In triple-stranded DNA, the third strand binds to a B-form DNA double helix by forming Hoogsteen base pairs or reversed Hoogsteen hydrogen bonds.
Carcinogenesis, also called oncogenesis or tumorigenesis, is the formation of a cancer, whereby normal cells are transformed into cancer cells. The process is characterized by changes at the cellular, genetic, and epigenetic levels and abnormal cell division. Cell division is a physiological process that occurs in almost all tissues and under a variety of circumstances. Normally, the balance between proliferation and programmed cell death, in the form of apoptosis, is maintained to ensure the integrity of tissues and organs. According to the prevailing accepted theory of carcinogenesis, the somatic mutation theory, mutations in DNA and epimutations that lead to cancer disrupt these orderly processes by interfering with the programming regulating the processes, upsetting the normal balance between proliferation and cell death. This results in uncontrolled cell division and the evolution of those cells by natural selection in the body. Only certain mutations lead to cancer whereas the majority of mutations do not.

DNA mismatch repair (MMR) is a system for recognizing and repairing erroneous insertion, deletion, and mis-incorporation of bases that can arise during DNA replication and recombination, as well as repairing some forms of DNA damage.
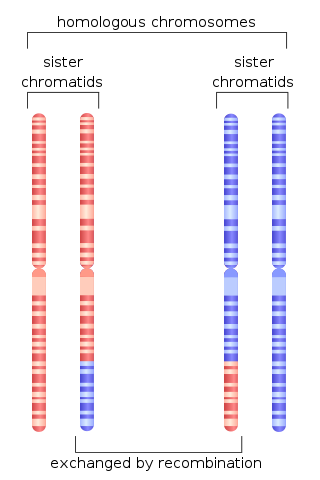
Homologous recombination is a type of genetic recombination in which genetic information is exchanged between two similar or identical molecules of double-stranded or single-stranded nucleic acids.

Werner syndrome ATP-dependent helicase, also known as DNA helicase, RecQ-like type 3, is an enzyme that in humans is encoded by the WRN gene. WRN is a member of the RecQ Helicase family. Helicase enzymes generally unwind and separate double-stranded DNA. These activities are necessary before DNA can be copied in preparation for cell division. Helicase enzymes are also critical for making a blueprint of a gene for protein production, a process called transcription. Further evidence suggests that Werner protein plays a critical role in repairing DNA. Overall, this protein helps maintain the structure and integrity of a person's DNA.

In biology, and especially in genetics, a mutant is an organism or a new genetic character arising or resulting from an instance of mutation, which is generally an alteration of the DNA sequence of the genome or chromosome of an organism. It is a characteristic that would not be observed naturally in a specimen. The term mutant is also applied to a virus with an alteration in its nucleotide sequence whose genome is in the nuclear genome. The natural occurrence of genetic mutations is integral to the process of evolution. The study of mutants is an integral part of biology; by understanding the effect that a mutation in a gene has, it is possible to establish the normal function of that gene.

DNA mismatch repair protein Mlh1 or MutL protein homolog 1 is a protein that in humans is encoded by the MLH1 gene located on chromosome 3. The gene is commonly associated with hereditary nonpolyposis colorectal cancer. Orthologs of human MLH1 have also been studied in other organisms including mouse and the budding yeast Saccharomyces cerevisiae.
Chromatin remodeling is the dynamic modification of chromatin architecture to allow access of condensed genomic DNA to the regulatory transcription machinery proteins, and thereby control gene expression. Such remodeling is principally carried out by 1) covalent histone modifications by specific enzymes, e.g., histone acetyltransferases (HATs), deacetylases, methyltransferases, and kinases, and 2) ATP-dependent chromatin remodeling complexes which either move, eject or restructure nucleosomes. Besides actively regulating gene expression, dynamic remodeling of chromatin imparts an epigenetic regulatory role in several key biological processes, egg cells DNA replication and repair; apoptosis; chromosome segregation as well as development and pluripotency. Aberrations in chromatin remodeling proteins are found to be associated with human diseases, including cancer. Targeting chromatin remodeling pathways is currently evolving as a major therapeutic strategy in the treatment of several cancers.

Mismatch repair endonuclease PMS2 is an enzyme that in humans is encoded by the PMS2 gene.
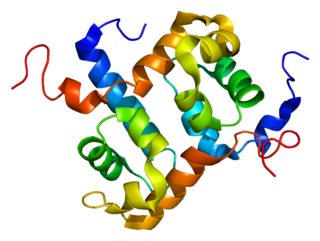
ERCC4 is a protein designated as DNA repair endonuclease XPF that in humans is encoded by the ERCC4 gene. Together with ERCC1, ERCC4 forms the ERCC1-XPF enzyme complex that participates in DNA repair and DNA recombination.

Probable helicase senataxin is an enzyme that in humans is encoded by the SETX gene.
The DNA damage theory of aging proposes that aging is a consequence of unrepaired accumulation of naturally occurring DNA damage. Damage in this context is a DNA alteration that has an abnormal structure. Although both mitochondrial and nuclear DNA damage can contribute to aging, nuclear DNA is the main subject of this analysis. Nuclear DNA damage can contribute to aging either indirectly or directly.

Cancer epigenetics is the study of epigenetic modifications to the DNA of cancer cells that do not involve a change in the nucleotide sequence, but instead involve a change in the way the genetic code is expressed. Epigenetic mechanisms are necessary to maintain normal sequences of tissue specific gene expression and are crucial for normal development. They may be just as important, if not even more important, than genetic mutations in a cell's transformation to cancer. The disturbance of epigenetic processes in cancers, can lead to a loss of expression of genes that occurs about 10 times more frequently by transcription silencing than by mutations. As Vogelstein et al. points out, in a colorectal cancer there are usually about 3 to 6 driver mutations and 33 to 66 hitchhiker or passenger mutations. However, in colon tumors compared to adjacent normal-appearing colonic mucosa, there are about 600 to 800 heavily methylated CpG islands in the promoters of genes in the tumors while these CpG islands are not methylated in the adjacent mucosa. Manipulation of epigenetic alterations holds great promise for cancer prevention, detection, and therapy. In different types of cancer, a variety of epigenetic mechanisms can be perturbed, such as the silencing of tumor suppressor genes and activation of oncogenes by altered CpG island methylation patterns, histone modifications, and dysregulation of DNA binding proteins. There are several medications which have epigenetic impact, that are now used in a number of these diseases.
Chromosomal instability (CIN) is a type of genomic instability in which chromosomes are unstable, such that either whole chromosomes or parts of chromosomes are duplicated or deleted. More specifically, CIN refers to the increase in rate of addition or loss of entire chromosomes or sections of them. The unequal distribution of DNA to daughter cells upon mitosis results in a failure to maintain euploidy leading to aneuploidy. In other words, the daughter cells do not have the same number of chromosomes as the cell they originated from. Chromosomal instability is the most common form of genetic instability and cause of aneuploidy.
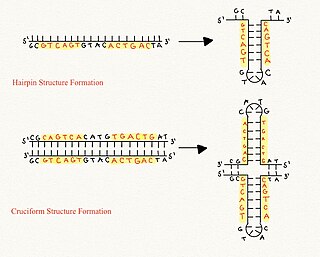
Cruciform DNA is a form of non-B DNA, or an alternative DNA structure. The formation of cruciform DNA requires the presence of palindromes called inverted repeat sequences. These inverted repeats contain a sequence of DNA in one strand that is repeated in the opposite direction on the other strand. As a result, inverted repeats are self-complementary and can give rise to structures such as hairpins and cruciforms. Cruciform DNA structures require at least a six nucleotide sequence of inverted repeats to form a structure consisting of a stem, branch point and loop in the shape of a cruciform, stabilized by negative DNA supercoiling.
References
- ↑ Darmon, E; Leach, DRF (2014). "Bacterial Genome Instability". Microbiol. Mol. Biol. Rev. 78 (1): 1–39. doi:10.1128/MMBR.00035-13. PMC 3957733 . PMID 24600039.
- ↑ Schmitt, MW; Prindle, MJ; Loeb, LA (2012). "Implications of genetic heterogeneity in cancer". Ann N Y Acad Sci. 1267 (1): 110–116. Bibcode:2012NYASA1267..110S. doi:10.1111/j.1749-6632.2012.06590.x. PMC 3674777 . PMID 22954224.
- ↑ Møller, P (2005). "Genotoxicity of environmental agents assessed by the alkaline comet assay". Basic Clin Pharmacol Toxicol. 96 (Suppl 1): 1–42. PMID 15859009.
- ↑ Keightley PD (February 2012). "Rates and fitness consequences of new mutations in humans". Genetics. 190 (2): 295–304. doi:10.1534/genetics.111.134668. PMC 3276617 . PMID 22345605.
- ↑ Aguilera, A; Klein, H. L. (Aug 1998). "Genetic control of intrachromosomal recombination in Saccharomyces cerevisiae. I. Isolation and genetic characterization of hyper-recombination mutations". Genetics. 4 (4): 779–790.
- ↑ Cobb, J. A. (Dec 2005). "Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations". Genes & Development. 19 (24): 3055–3069. doi:10.1101/gad.361805. PMC 1315408 . PMID 16357221.
- ↑ Cortes-Ledesma, Felipe; Aguilera, Andres (Sep 2006). "Double-strand breaks arising from replication through a nick are repaired by cohesin-dependent sister-chromatid exchange". EMBO Reports. 7 (9): 919–926. doi:10.1038/sj.embor.7400774. PMC 1559660 . PMID 16888651.
- ↑ Weinert, T. A.; Hartwell, L. H. (May 1993). "Cell cycle arrest of cdc mutants and specificity of the RAD9 checkpoint". Genetics. 134 (1): 63–80. doi:10.1093/genetics/134.1.63. PMC 1205445 . PMID 8514150.
- ↑ Durkin, Sandra G.; Glover, Thomas W. (Dec 2007). "Chromosome Fragile Sites". Annual Review of Genetics. 41 (1): 169–192. doi:10.1146/annurev.genet.41.042007.165900. PMID 17608616.
- ↑ Grabczyk, E.; Mancuso, M.; Sammarco, M. C. (Aug 2007). "A persistent RNA-DNA hybrid formed by transcription of the Friedreich ataxia triplet repeat in live bacteria, and by T7 RNAP in vitro". Nucleic Acids Research. 35 (16): 5351–5359. doi:10.1093/nar/gkm589. PMC 2018641 . PMID 17693431.
- ↑ Trautinger, Brigitte W.; Jaktaji, Razieh P.; Rusakova, Ekaterina; Lloyd, Robert G. (July 2005). "RNA Polymerase Modulators and DNA Repair Activities Resolve Conflicts between DNA Replication and Transcription". Molecular Cell. 19 (2): 247–258. doi: 10.1016/j.molcel.2005.06.004 . PMID 16039593.
- ↑ Schrader, Carol E.; Guikema, Jeroen E. J.; Linehan, Erin K.; Selsing, Erik; Stavnezer, Janet (Nov 2007). "Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair". Journal of Immunology. 179 (9): 6064–6071. doi: 10.4049/jimmunol.179.9.6064 . PMID 17947680.
- ↑ Subba Rao, K (2007). "Mechanisms of disease: DNA repair defects and neurological disease". Nat Clin Pract Neurol. 3 (3): 162–72. doi:10.1038/ncpneuro0448. PMID 17342192. S2CID 12930631.
- ↑ Jeppesen, DK; Bohr, VA; Stevnsner, T (2011). "DNA repair deficiency in neurodegeneration". Prog Neurobiol. 94 (2): 166–200. doi:10.1016/j.pneurobio.2011.04.013. PMC 3123739 . PMID 21550379.
- ↑ Corcos, D. (2012), "Unbalanced replication as a major source of genetic instability in cancer cells", American Journal of Blood Research, 2 (3): 160–9, PMC 3484411 , PMID 23119227
- ↑ Storchova, Z.; Pellman, D. (2004), "From polyploidy to aneuploidy, genome instability and cancer", Nat Rev Mol Cell Biol, 5 (1): 45–54, doi:10.1038/nrm1276, PMID 14708009, S2CID 11985415
- 1 2 3 Vogelstein B; Papadopoulos N; Velculescu VE; Zhou S; Diaz LA; Kinzler KW (March 2013). "Cancer genome landscapes". Science. 339 (6127): 1546–58. Bibcode:2013Sci...339.1546V. doi:10.1126/science.1235122. PMC 3749880 . PMID 23539594.
- ↑ Nowak, M. A.; Komarova, N. L.; Sengupta, A.; Jallepalli, P.V.; Shih, I.M.; Vogelstein, B.; Lengauer, C. (2002), "The role of chromosomal instability in tumor initiation", Proc. Natl. Acad. Sci. USA, 99 (25): 16226–31, Bibcode:2002PNAS...9916226N, doi: 10.1073/pnas.202617399 , PMC 138593 , PMID 12446840
- ↑ Kinzler, K. W.; Vogelstein, B. (April 1997), "Cancer-susceptibility genes. Gatekeepers and caretakers", Nature, 386 (6627): 761–3, doi: 10.1038/386761a0 , PMID 9126728
- ↑ Cahill, D. P.; Kinzler, K. W.; Vogelstein, B.; Lengauer, C. (1999), "Genetic instability and darwinian selection in tumours", Trends Cell Biol., 9 (12): M57–M60, doi: 10.1016/S0168-9525(99)01874-0 , PMID 10611684
- ↑ Hui, T.; Zhen, G.; HuiZhong, L.; BaoFu, Z.; Gang, W.; Qing, Z.; DongSheng, P.; JunNian, Z. (2015), "DNA damage response – A double-edged sword in cancer prevention and cancer therapy", Cancer Letters, 358 (1): 8–16, doi:10.1016/j.canlet.2014.12.038, PMID 25528631
- ↑ Lander ES; Linton LM; Birren B; Nusbaum C; Zody MC; Baldwin J; Devon K; Dewar K; Doyle M; FitzHugh W; et al. (February 2001). "Initial sequencing and analysis of the human genome" (PDF). Nature. 409 (6822): 860–921. Bibcode:2001Natur.409..860L. doi: 10.1038/35057062 . PMID 11237011.
- ↑ Roach JC; Glusman G; Smit AF; et al. (April 2010). "Analysis of genetic inheritance in a family quartet by whole-genome sequencing". Science. 328 (5978): 636–9. Bibcode:2010Sci...328..636R. doi:10.1126/science.1186802. PMC 3037280 . PMID 20220176.
- ↑ Campbell CD; Chong JX; Malig M; et al. (November 2012). "Estimating the human mutation rate using autozygosity in a founder population". Nat. Genet. 44 (11): 1277–81. doi:10.1038/ng.2418. PMC 3483378 . PMID 23001126.
- ↑ Cunningham, FH; Fiebelkorn, S; Johnson, M; Meredith, C (2011). "A novel application of the Margin of Exposure approach: segregation of tobacco smoke toxicants". Food Chem Toxicol. 49 (11): 2921–2933. doi:10.1016/j.fct.2011.07.019. PMID 21802474.
- ↑ Cuozzo, C; Porcellini, A; Angrisano, T; Morano, A; Lee, B; Di Pardo, A; Messina, S; Iuliano, R; Fusco, A; Santillo, MR; Muller, MT; Chiariotti, L; Gottesman, ME; Avvedimento, EV (2007). "DNA damage, homology-directed repair, and DNA methylation". PLOS Genet. 3 (7): e110. doi: 10.1371/journal.pgen.0030110 . PMC 1913100 . PMID 17616978.
- ↑ O'Hagan, HM; Mohammad, HP; Baylin, SB (2008). "Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island". PLOS Genet. 4 (8): e1000155. doi: 10.1371/journal.pgen.1000155 . PMC 2491723 . PMID 18704159.
- ↑ Gottschalk, AJ; Timinszky, G; Kong, SE; Jin, J; Cai, Y; Swanson, SK; Washburn, MP; Florens, L; Ladurner, AG; Conaway, JW; Conaway, RC (2009). "Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler". Proc Natl Acad Sci U S A. 106 (33): 13770–4. Bibcode:2009PNAS..10613770G. doi: 10.1073/pnas.0906920106 . PMC 2722505 . PMID 19666485.
- ↑ Yost SE; Smith EN; Schwab RB; Bao L; Jung H; Wang X; Voest E; Pierce JP; Messer K; Parker BA; Harismendy O; Frazer KA (August 2012). "Identification of high-confidence somatic mutations in whole genome sequence of formalin-fixed breast cancer specimens". Nucleic Acids Res. 40 (14): e107. doi:10.1093/nar/gks299. PMC 3413110 . PMID 22492626.
- ↑ Berger MF; Hodis E; Heffernan TP; Deribe YL; Lawrence MS; Protopopov A; Ivanova E; Watson IR; Nickerson E; Ghosh P; Zhang H; Zeid R; Ren X; Cibulskis K; Sivachenko AY; Wagle N; Sucker A; Sougnez C; Onofrio R; Ambrogio L; Auclair D; Fennell T; Carter SL; Drier Y; Stojanov P; Singer MA; Voet D; Jing R; Saksena G; Barretina J; Ramos AH; Pugh TJ; Stransky N; Parkin M; Winckler W; Mahan S; Ardlie K; Baldwin J; Wargo J; Schadendorf D; Meyerson M; Gabriel SB; Golub TR; Wagner SN; Lander ES; Getz G; Chin L; Garraway LA (May 2012). "Melanoma genome sequencing reveals frequent PREX2 mutations". Nature. 485 (7399): 502–6. Bibcode:2012Natur.485..502B. doi:10.1038/nature11071. PMC 3367798 . PMID 22622578.
- ↑ Narayanan L; Fritzell JA; Baker SM; Liskay RM; Glazer PM (April 1997). "Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2". Proc. Natl. Acad. Sci. U.S.A. 94 (7): 3122–7. Bibcode:1997PNAS...94.3122N. doi: 10.1073/pnas.94.7.3122 . PMC 20332 . PMID 9096356.
- ↑ Hegan DC; Narayanan L; Jirik FR; Edelmann W; Liskay RM; Glazer PM (December 2006). "Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6". Carcinogenesis. 27 (12): 2402–8. doi:10.1093/carcin/bgl079. PMC 2612936 . PMID 16728433.
- ↑ Tutt AN; van Oostrom CT; Ross GM; van Steeg H; Ashworth A (March 2002). "Disruption of Brca2 increases the spontaneous mutation rate in vivo: synergism with ionizing radiation". EMBO Rep. 3 (3): 255–60. doi:10.1093/embo-reports/kvf037. PMC 1084010 . PMID 11850397.
- ↑ German, J (Mar 1969). "Bloom's syndrome. I. Genetical and clinical observations in the first twenty-seven patients". Am J Hum Genet. 21 (2): 196–227. PMC 1706430 . PMID 5770175.
- ↑ Halford S; Rowan A; Sawyer E; Talbot I; Tomlinson I (June 2005). "O(6)-methylguanine methyltransferase in colorectal cancers: detection of mutations, loss of expression, and weak association with G:C>A:T transitions". Gut. 54 (6): 797–802. doi:10.1136/gut.2004.059535. PMC 1774551 . PMID 15888787.
- ↑ Truninger, K; Menigatti, M; Luz, J; Russell, A; Haider, R; Gebbers, JO; Bannwart, F; Yurtsever, H; Neuweiler, J; Riehle, HM; Cattaruzza, MS; Heinimann, K; Schär, P; Jiricny, J; Marra, G (2005). "Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer". Gastroenterology. 128 (5): 1160–1171. doi: 10.1053/j.gastro.2005.01.056 . PMID 15887099.
- ↑ Valeri, N; Gasparini, P; Fabbri, M; Braconi, C; Veronese, A; Lovat, F; Adair, B; Vannini, I; Fanini, F; Bottoni, A; Costinean, S; Sandhu, SK; Nuovo, GJ; Alder, H; Gafa, R; Calore, F; Ferracin, M; Lanza, G; Volinia, S; Negrini, M; Mcllhatton, MA; Amadori, D; Fishel, R; Croce, CM (2010). "Modulation of mismatch repair and genomic stability by miR-155". Proc Natl Acad Sci USA. 107 (15): 6982–6987. Bibcode:2010PNAS..107.6982V. doi: 10.1073/pnas.1002472107 . PMC 2872463 . PMID 20351277.
- ↑ Facista, A; Nguyen, H; Lewis, C; Prasad, AR; Ramsey, L; Zaitlin, B; Nfonsam, V; Krouse, RS; Bernstein, H; Payne, CM; Stern, S; Oatman, N; Banerjee, B; Bernstein, C (2012). "Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer". Genome Integr. 3 (1): 3. doi: 10.1186/2041-9414-3-3 . PMC 3351028 . PMID 22494821.
- ↑ Zheng, Jie (Nov 2013). "Oncogenic chromosomal translocations and human cancer (Review)". Oncology Reports. 30 (5): 2011–2019. doi: 10.3892/or.2013.2677 . PMID 23970180.
- ↑ Ramiro, Almudena; San-Marin, Bernardo Reina; McBride, Kevin; Jankovic, Mila; Barreto, Vasco; Nussenzweig, Andre; Nussenzweig, Michel C. (2007). Advances in Immunology. Elsevier. pp. 75–107. ISBN 978-0-12-373706-9.