
Proteins are large biomolecules and macromolecules that comprise one or more long chains of amino acid residues. Proteins perform a vast array of functions within organisms, including catalysing metabolic reactions, DNA replication, responding to stimuli, providing structure to cells and organisms, and transporting molecules from one location to another. Proteins differ from one another primarily in their sequence of amino acids, which is dictated by the nucleotide sequence of their genes, and which usually results in protein folding into a specific 3D structure that determines its activity.

Protein folding is the physical process by which a protein, after synthesis by a ribosome as a linear chain of amino acids, changes from an unstable random coil into a more ordered three-dimensional structure. This structure permits the protein to become biologically functional.

Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; it is important in medicine and biotechnology.
An epitope, also known as antigenic determinant, is the part of an antigen that is recognized by the immune system, specifically by antibodies, B cells, or T cells. The part of an antibody that binds to the epitope is called a paratope. Although epitopes are usually non-self proteins, sequences derived from the host that can be recognized are also epitopes.

Protein structure is the three-dimensional arrangement of atoms in an amino acid-chain molecule. Proteins are polymers – specifically polypeptides – formed from sequences of amino acids, which are the monomers of the polymer. A single amino acid monomer may also be called a residue, which indicates a repeating unit of a polymer. Proteins form by amino acids undergoing condensation reactions, in which the amino acids lose one water molecule per reaction in order to attach to one another with a peptide bond. By convention, a chain under 30 amino acids is often identified as a peptide, rather than a protein. To be able to perform their biological function, proteins fold into one or more specific spatial conformations driven by a number of non-covalent interactions, such as hydrogen bonding, ionic interactions, Van der Waals forces, and hydrophobic packing. To understand the functions of proteins at a molecular level, it is often necessary to determine their three-dimensional structure. This is the topic of the scientific field of structural biology, which employs techniques such as X-ray crystallography, NMR spectroscopy, cryo-electron microscopy (cryo-EM) and dual polarisation interferometry, to determine the structure of proteins.
In biochemistry, a conformational change is a change in the shape of a macromolecule, often induced by environmental factors.

In molecular biology, a protein domain is a region of a protein's polypeptide chain that is self-stabilizing and that folds independently from the rest. Each domain forms a compact folded three-dimensional structure. Many proteins consist of several domains, and a domain may appear in a variety of different proteins. Molecular evolution uses domains as building blocks and these may be recombined in different arrangements to create proteins with different functions. In general, domains vary in length from between about 50 amino acids up to 250 amino acids in length. The shortest domains, such as zinc fingers, are stabilized by metal ions or disulfide bridges. Domains often form functional units, such as the calcium-binding EF hand domain of calmodulin. Because they are independently stable, domains can be "swapped" by genetic engineering between one protein and another to make chimeric proteins.
In molecular biology, proteins are generally thought to adopt unique structures determined by their amino acid sequences. However, proteins are not strictly static objects, but rather populate ensembles of conformations. Transitions between these states occur on a variety of length scales and time scales , and have been linked to functionally relevant phenomena such as allosteric signaling and enzyme catalysis.

In molecular biology short linear motifs (SLiMs), linear motifs or minimotifs are short stretches of protein sequence that mediate protein–protein interaction.
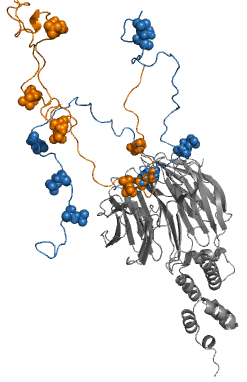
Fuzzy complexes are protein complexes, where structural ambiguity or multiplicity exists and is required for biological function. Alteration, truncation or removal of conformationally ambiguous regions impacts the activity of the corresponding complex. Fuzzy complexes are generally formed by intrinsically disordered proteins. Structural multiplicity usually underlies functional multiplicity of protein complexes following a fuzzy logic. Distinct binding modes of the nucleosome are also regarded as a special case of fuzziness.

In molecular biology, protein fold classes are broad categories of protein tertiary structure topology. They describe groups of proteins that share similar amino acid and secondary structure proportions. Each class contains multiple, independent protein superfamilies.

Fast parallel proteolysis (FASTpp) is a method to determine the thermostability of proteins by measuring which fraction of protein resists rapid proteolytic digestion.
Molecular recognition features (MoRFs) are small intrinsically disordered regions in proteins that undergo a disorder-to-order transition upon binding to their partners. MoRFs are implicated in protein-protein interactions, which serve as the initial step in molecular recognition. MoRFs are disordered prior to binding to their partners, whereas they form a common 3D structure after interacting with their partners. As MoRF regions tend to resemble disordered proteins with some characteristics of ordered proteins, they can be classified as existing in an extended semi-disordered state.
A protein superfamily is the largest grouping (clade) of proteins for which common ancestry can be inferred. Usually this common ancestry is inferred from structural alignment and mechanistic similarity, even if no sequence similarity is evident. Sequence homology can then be deduced even if not apparent. Superfamilies typically contain several protein families which show sequence similarity within each family. The term protein clan is commonly used for protease and glycosyl hydrolases superfamilies based on the MEROPS and CAZy classification systems.

In computational chemistry, conformational ensembles, also known as structural ensembles, are experimentally constrained computational models describing the structure of intrinsically unstructured proteins. Such proteins are flexible in nature, lacking a stable tertiary structure, and therefore cannot be described with a single structural representation. The techniques of ensemble calculation are relatively new on the field of structural biology, and are still facing certain limitations that need to be addressed before it will become comparable to classical structural description methods such as biological macromolecular crystallography.

Proline-rich protein 30 is a protein in humans that is encoded for by the PRR30 gene. PRR30 is a member in the family of Proline-rich proteins characterized by their intrinsic lack of structure. Copy number variations in the PRR30 gene have been associated with an increased risk for neurofibromatosis.

An array of protein tandem repeats is defined as several adjacent copies having the same or similar sequence motifs. These periodic sequences are generated by internal duplications in both coding and non-coding genomic sequences. Repetitive units of protein tandem repeats are considerably diverse, ranging from the repetition of a single amino acid to domains of 100 or more residues.
Low complexity regions (LCRs) in protein sequences, also defined in some contexts as compositionally biased regions (CBRs), are regions in protein sequences that differ from the composition and complexity of most proteins that is normally associated with globular structure. LCRs have different properties from normal regions regarding structure, function and evolution.
The dark proteome is defined as proteins with no defined three-dimensional structure. It can not be detected or analyzed with the use of homologous modeling or analytical quantification for the molecular conformation is unknown. Dark proteins are mostly composed of unknown unknowns.
LLPS often involves sequence regions that have unique functional characteristics, as well as the presence of prion-like and RNA-binding domains. Nowadays there are just a few methods to predict the propensity of a protein to drive LLPS. The range of biological mechanisms involved in LLPS, the limited knowledge about these mechanisms and the important context-dependent component of LLPS make this problem challenging. In the last years, despite the advances in this field, just few predictors, specific for LLPS, have been developed, trying to understand the relationship between protein sequence properties and the capability to drive LLPS. Here we will revise the state-of-the-art LLPS sequence-based predictors, briefly introducing them and explaining which are the individual protein characteristics that they identify in the context of LLPS.



