
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences, such as calculating the distance cost between strings in a natural language or in financial data.
In bioinformatics, sequence clustering algorithms attempt to group biological sequences that are somehow related. The sequences can be either of genomic, "transcriptomic" (ESTs) or protein origin. For proteins, homologous sequences are typically grouped into families. For EST data, clustering is important to group sequences originating from the same gene before the ESTs are assembled to reconstruct the original mRNA.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.
BioJava is an open-source software project dedicated to provide Java tools to process biological data. BioJava is a set of library functions written in the programming language Java for manipulating sequences, protein structures, file parsers, Common Object Request Broker Architecture (CORBA) interoperability, Distributed Annotation System (DAS), access to AceDB, dynamic programming, and simple statistical routines. BioJava supports a huge range of data, starting from DNA and protein sequences to the level of 3D protein structures. The BioJava libraries are useful for automating many daily and mundane bioinformatics tasks such as to parsing a Protein Data Bank (PDB) file, interacting with Jmol and many more. This application programming interface (API) provides various file parsers, data models and algorithms to facilitate working with the standard data formats and enables rapid application development and analysis.

UniProt is a freely accessible database of protein sequence and functional information, many entries being derived from genome sequencing projects. It contains a large amount of information about the biological function of proteins derived from the research literature. It is maintained by the UniProt consortium, which consists of several European bioinformatics organisations and a foundation from Washington, DC, United States.
Clustal is a series of widely used computer programs used in bioinformatics for multiple sequence alignment. There have been many versions of Clustal over the development of the algorithm that are listed below. The analysis of each tool and its algorithm are also detailed in their respective categories. Available operating systems listed in the sidebar are a combination of the software availability and may not be supported for every current version of the Clustal tools. Clustal Omega has the widest variety of operating systems out of all the Clustal tools.

Ensembl genome database project is a scientific project at the European Bioinformatics Institute, which provides a centralized resource for geneticists, molecular biologists and other researchers studying the genomes of our own species and other vertebrates and model organisms. Ensembl is one of several well known genome browsers for the retrieval of genomic information.
The European Bioinformatics Institute (EMBL-EBI) is an intergovernmental organization (IGO) which, as part of the European Molecular Biology Laboratory (EMBL) family, focuses on research and services in bioinformatics. It is located on the Wellcome Genome Campus in Hinxton near Cambridge, and employs over 600 full-time equivalent (FTE) staff. Institute leaders such as Rolf Apweiler, Alex Bateman, Ewan Birney, and Guy Cochrane, an adviser on the National Genomics Data Center Scientific Advisory Board, serve as part of the international research network of the BIG Data Center at the Beijing Institute of Genomics.

Pfam is a database of protein families that includes their annotations and multiple sequence alignments generated using hidden Markov models. The most recent version, Pfam 35.0, was released in November 2021 and contains 19,632 families.

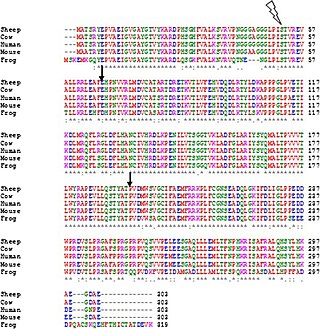
Multiple sequence alignment (MSA) may refer to the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a linkage and are descended from a common ancestor. From the resulting MSA, sequence homology can be inferred and phylogenetic analysis can be conducted to assess the sequences' shared evolutionary origins. Visual depictions of the alignment as in the image at right illustrate mutation events such as point mutations that appear as differing characters in a single alignment column, and insertion or deletion mutations that appear as hyphens in one or more of the sequences in the alignment. Multiple sequence alignment is often used to assess sequence conservation of protein domains, tertiary and secondary structures, and even individual amino acids or nucleotides.
InterPro is a database of protein families, protein domains and functional sites in which identifiable features found in known proteins can be applied to new protein sequences in order to functionally characterise them.

In bioinformatics, MAFFT is a program used to create multiple sequence alignments of amino acid or nucleotide sequences. Published in 2002, the first version of MAFFT used an algorithm based on progressive alignment, in which the sequences were clustered with the help of the Fast Fourier Transform. Subsequent versions of MAFFT have added other algorithms and modes of operation, including options for faster alignment of large numbers of sequences, higher accuracy alignments, alignment of non-coding RNA sequences, and the addition of new sequences to existing alignments.
MUltiple Sequence Comparison by Log-Expectation (MUSCLE) is computer software for multiple sequence alignment of protein and nucleotide sequences. It is licensed as public domain. The method was published by Robert C. Edgar in two papers in 2004. The first paper, published in Nucleic Acids Research, introduced the sequence alignment algorithm. The second paper, published in BMC Bioinformatics, presented more technical details.

UTOPIA is a suite of free tools for visualising and analysing bioinformatics data. Based on an ontology-driven data model, it contains applications for viewing and aligning protein sequences, rendering complex molecular structures in 3D, and for finding and using resources such as web services and data objects. There are two major components, the protein analysis suite and UTOPIA documents.
The Viral Bioinformatics Resource Center (VBRC) is an online resource providing access to a database of curated viral genomes and a variety of tools for bioinformatic genome analysis. This resource was one of eight BRCs funded by NIAID with the goal of promoting research against emerging and re-emerging pathogens, particularly those seen as potential bioterrorism threats. The VBRC is now supported by Dr. Chris Upton at the University of Victoria.

UGENE is computer software for bioinformatics. It works on personal computer operating systems such as Windows, macOS, or Linux. It is released as free and open-source software, under a GNU General Public License (GPL) version 2.

HMMER is a free and commonly used software package for sequence analysis written by Sean Eddy. Its general usage is to identify homologous protein or nucleotide sequences, and to perform sequence alignments. It detects homology by comparing a profile-HMM to either a single sequence or a database of sequences. Sequences that score significantly better to the profile-HMM compared to a null model are considered to be homologous to the sequences that were used to construct the profile-HMM. Profile-HMMs are constructed from a multiple sequence alignment in the HMMER package using the hmmbuild program. The profile-HMM implementation used in the HMMER software was based on the work of Krogh and colleagues. HMMER is a console utility ported to every major operating system, including different versions of Linux, Windows, and Mac OS.

Teresa K. Attwood is a Professor of Bioinformatics in the Department of Computer Science and School of Biological Sciences at the University of Manchester and a visiting fellow at the European Bioinformatics Institute (EMBL-EBI). She held a Royal Society University Research Fellowship at University College London (UCL) from 1993 to 1999 and at the University of Manchester from 1999 to 2002.

Alexander George Bateman is a computational biologist and Head of Protein Sequence Resources at the European Bioinformatics Institute (EBI), part of the European Molecular Biology Laboratory (EMBL) in Cambridge, UK. He has led the development of the Pfam biological database and introduced the Rfam database of RNA families. He has also been involved in the use of Wikipedia for community-based annotation of biological databases.
