
A brain tumor occurs when abnormal cells form within the brain. There are two main types of tumors: malignant (cancerous) tumors and benign (non-cancerous) tumors. These can be further classified as primary tumors, which start within the brain, and secondary tumors, which most commonly have spread from tumors located outside the brain, known as brain metastasis tumors. All types of brain tumors may produce symptoms that vary depending on the size of the tumor and the part of the brain that is involved. Where symptoms exist, they may include headaches, seizures, problems with vision, vomiting and mental changes. Other symptoms may include difficulty walking, speaking, with sensations, or unconsciousness.

Retinoblastoma (Rb) is a rare form of cancer that rapidly develops from the immature cells of a retina, the light-detecting tissue of the eye. It is the most common primary malignant intraocular cancer in children, and it is almost exclusively found in young children.

Oligodendrogliomas are a type of glioma that are believed to originate from the oligodendrocytes of the brain or from a glial precursor cell. They occur primarily in adults but are also found in children.

Small-cell carcinoma is a type of highly malignant cancer that most commonly arises within the lung, although it can occasionally arise in other body sites, such as the cervix, prostate, and gastrointestinal tract. Compared to non-small cell carcinoma, small cell carcinoma is more aggressive, with a shorter doubling time, higher growth fraction, and earlier development of metastases.
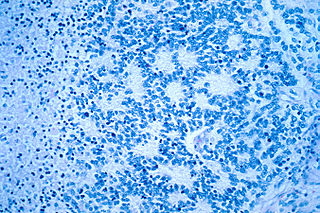
Neuroblastoma (NB) is a type of cancer that forms in certain types of nerve tissue. It most frequently starts from one of the adrenal glands but can also develop in the head, neck, chest, abdomen, or spine. Symptoms may include bone pain, a lump in the abdomen, neck, or chest, or a painless bluish lump under the skin.
Adjuvant therapy, also known as adjunct therapy, adjuvant care, or augmentation therapy, is a therapy that is given in addition to the primary or initial therapy to maximize its effectiveness. The surgeries and complex treatment regimens used in cancer therapy have led the term to be used mainly to describe adjuvant cancer treatments. An example of such adjuvant therapy is the additional treatment usually given after surgery where all detectable disease has been removed, but where there remains a statistical risk of relapse due to the presence of undetected disease. If known disease is left behind following surgery, then further treatment is not technically adjuvant.

A blastoma is a type of cancer, more common in children, that is caused by malignancies in precursor cells, often called blasts. Examples are nephroblastoma, medulloblastoma, and retinoblastoma. The suffix -blastoma is used to imply a tumor of primitive, incompletely differentiated cells, e.g., chondroblastoma is composed of cells resembling the precursor of chondrocytes.

Anaplastic thyroid cancer (ATC), also known as anaplastic thyroid carcinoma, is an aggressive form of thyroid cancer characterized by uncontrolled growth of cells in the thyroid gland. This form of cancer generally carries a very poor prognosis due to its aggressive behavior and resistance to cancer treatments. The cells of anaplastic thyroid cancer are highly abnormal and usually no longer resemble the original thyroid cells and have poor differentiation.

Primitive neuroectodermal tumor is a malignant (cancerous) neural crest tumor. It is a rare tumor, usually occurring in children and young adults under 25 years of age. The overall 5 year survival rate is about 53%.
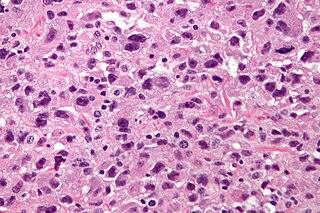
Undifferentiated pleomorphic sarcoma (UPS), also termed pleomorphic myofibrosarcoma, high-grade myofibroblastic sarcoma, and high-grade myofibrosarcoma, is characterized by the World Health Organization (WHO), 2020, as a rare, poorly differentiated neoplasm, i.e. an abnormal growth of cells that have an unclear identity and/or cell of origin. WHO classified it as one of the undifferentiated/unclassified sarcomas in the category of tumors of uncertain differentiation. Sarcomas are cancers known or thought to derive from mesenchymal stem cells that typically develop in bone, muscle, fat, blood vessels, lymphatic vessels, tendons, and ligaments. More than 70 sarcoma subtypes have been described. The UPS subtype of these sarcomas consists of tumor cells that are poorly differentiated and may appear as spindle-shaped cells, histiocytes, and giant cells. UPS is considered a diagnosis that defies formal sub-classification after thorough histologic, immunohistochemical, and ultrastructural examinations fail to identify the type of cells involved.

An atypical teratoid rhabdoid tumor (AT/RT) is a rare tumor usually diagnosed in childhood. Although usually a brain tumor, AT/RT can occur anywhere in the central nervous system (CNS), including the spinal cord. About 60% will be in the posterior cranial fossa. One review estimated 52% in the posterior fossa, 39% are supratentorial primitive neuroectodermal tumors (sPNET), 5% are in the pineal, 2% are spinal, and 2% are multifocal.
Alveolar rhabdomyosarcoma (ARMS) is a subtype of the rhabdomyosarcoma soft tissue cancer family whose lineage is from mesenchymal cells and are related to skeletal muscle cells. ARMS tumors resemble the alveolar tissue in the lungs. Tumor location varies from patient to patient, but is commonly found in the head and neck region, male and female urogenital tracts, the torso, and extremities. Two fusion proteins can be associated with ARMS, but are not necessary, PAX3-FKHR. and PAX7-FKHR. In children and adolescents ARMS accounts for about 1 percent of all malignancies, has an incidence rate of 1 per million, and most cases occur sporadically with no genetic predisposition. PAX3-FOXO1 is now known to drive cancer-promoting gene expression programs through creation of distant genetic elements called super enhancers.
Triple-negative breast cancer (TNBC) is any breast cancer that either lacks or shows low levels of estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) overexpression and/or gene amplification. Triple-negative is sometimes used as a surrogate term for basal-like.
The Ewing family of tumors (EFTs) is a group of small cell sarcomas including Ewing sarcoma of the bone, extra osseous Ewing tumors, and primitive neuroectodermal tumors. They are rare cancers, usually diagnosed in peoples' twenties. The sarcoma of bone is the most common of the variants. All forms are predisposed to metastasis and have had historically high rates of mortality. The family of tumors shares a common translocation mutation of the EWS gene on chromosome 22 to an ETS-type gene, most commonly the FLI1 gene. EFTs are highly malignant, with 5-year survival for patients with metastatic disease at 20%. The current standard of care includes resection, radiation, and chemotherapy.

Nodular lymphocyte predominant Hodgkin lymphoma (NLPHL) is a slow-growing CD20 positive form of Hodgkin lymphoma, a cancer of the immune system's B cells.
Virtual karyotype is the digital information reflecting a karyotype, resulting from the analysis of short sequences of DNA from specific loci all over the genome, which are isolated and enumerated. It detects genomic copy number variations at a higher resolution for level than conventional karyotyping or chromosome-based comparative genomic hybridization (CGH). The main methods used for creating virtual karyotypes are array-comparative genomic hybridization and SNP arrays.

A brain metastasis is a cancer that has metastasized (spread) to the brain from another location in the body and is therefore considered a secondary brain tumor. The metastasis typically shares a cancer cell type with the original site of the cancer. Metastasis is the most common cause of brain cancer, as primary tumors that originate in the brain are less common. The most common sites of primary cancer which metastasize to the brain are lung, breast, colon, kidney, and skin cancer. Brain metastases can occur months or even years after the original or primary cancer is treated. Brain metastases have a poor prognosis for cure, but modern treatments allow patients to live months and sometimes years after the diagnosis.
Pediatric ependymomas are similar in nature to the adult form of ependymoma in that they are thought to arise from radial glial cells lining the ventricular system. However, they differ from adult ependymomas in which genes and chromosomes are most often affected, the region of the brain they are most frequently found in, and the prognosis of the patients. Children with certain hereditary diseases, such as neurofibromatosis type II (NF2), have been found to be more frequently afflicted with this class of tumors, but a firm genetic link remains to be established. Symptoms associated with the development of pediatric ependymomas are varied, much like symptoms for a number of other pediatric brain tumors including vomiting, headache, irritability, lethargy, and changes in gait. Although younger children and children with invasive tumor types generally experience less favorable outcomes, total removal of the tumors is the most conspicuous prognostic factor for both survival and relapse.

A central nervous system primitive neuroectodermal tumor, often abbreviated as PNET, supratentorial PNET, or CNS-PNET, is one of the 3 types of embryonal central nervous system tumors. It is considered an embryonal tumor because it arises from cells partially differentiated or still undifferentiated from birth. Those cells are usually neuroepithelial cells, stem cells destined to turn into glia or neurons. It can occur anywhere within the spinal cord and cerebrum and can have multiple sites of origins, with a high probability of metastasis through cerebrospinal fluid (CSF).
Embryonal tumor with multilayered rosettes (ETMR) is an embryonal central nervous system tumor. It is considered an embryonal tumor because it arises from cells partially differentiated or still undifferentiated from birth, usually neuroepithelial cells, stem cells destined to turn into glia or neurons. It can occur anywhere within the brain and can have multiple sites of origins, with a high probability of metastasis through cerebrospinal fluid (CSF). Metastases outside the central nervous system have been reported, but remain rare.


