A tumor suppressor gene (TSG), or anti-oncogene, is a gene that regulates a cell during cell division and replication. If the cell grows uncontrollably, it will result in cancer. When a tumor suppressor gene is mutated, it results in a loss or reduction in its function. In combination with other genetic mutations, this could allow the cell to grow abnormally. The loss of function for these genes may be even more significant in the development of human cancers, compared to the activation of oncogenes.

Retinitis pigmentosa (RP) is a genetic disorder of the eyes that causes loss of vision. Symptoms include trouble seeing at night and decreasing peripheral vision. As peripheral vision worsens, people may experience "tunnel vision". Complete blindness is uncommon. Onset of symptoms is generally gradual and often begins in childhood.
Norrie disease is a rare X-linked recessive genetic disorder that primarily affects the eyes and almost always leads to blindness. It is caused by mutations in the Norrin cystine knot growth factor gene, also referred to as Norrie Disease Pseudoglioma (NDP) gene. Norrie disease manifests with vision impairment either at birth, or within a few weeks of life, following an ocular event like retinal detachment and is progressive through childhood and adolescence. It generally begins with retinal degeneration, which occurs before birth and results in blindness at birth (congenital) or early infancy, usually by 3 months of age.

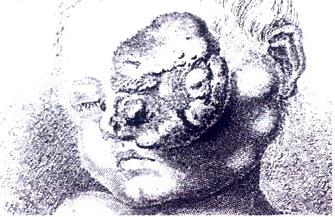
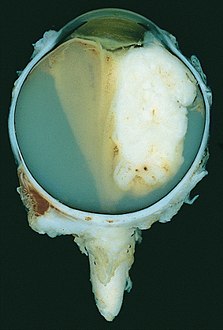
A blastoma is a type of cancer, more common in children, that is caused by malignancies in precursor cells, often called blasts. Examples are nephroblastoma, medulloblastoma, and retinoblastoma. The suffix -blastoma is used to imply a tumor of primitive, incompletely differentiated cells, e.g., chondroblastoma is composed of cells resembling the precursor of chondrocytes.

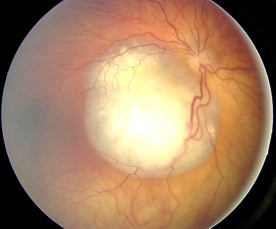
Uveal melanoma is a type of eye cancer in the uvea of the eye. It is traditionally classed as originating in the iris, choroid, and ciliary body, but can also be divided into class I and class II. Symptoms include blurred vision, loss of vision or photopsia, but there may be no symptoms.

Choroideremia is a rare, X-linked recessive form of hereditary retinal degeneration that affects roughly 1 in 50,000 males. The disease causes a gradual loss of vision, starting with childhood night blindness, followed by peripheral vision loss and progressing to loss of central vision later in life. Progression continues throughout the individual's life, but both the rate of change and the degree of visual loss are variable among those affected, even within the same family.
Stargardt disease is the most common inherited single-gene retinal disease. In terms of the first description of the disease, it follows an autosomal recessive inheritance pattern, which has been later linked to bi-allelic ABCA4 gene variants (STGD1). However, there are Stargardt-like diseases with mimicking phenotypes that are referred to as STGD3 and STGD4, and have a autosomal dominant inheritance due to defects with ELOVL4 or PROM1 genes, respectively. It is characterized by macular degeneration that begins in childhood, adolescence or adulthood, resulting in progressive loss of vision.

An eye neoplasm is a tumor of the eye. A rare type of tumor, eye neoplasms can affect all parts of the eye, and can either be benign or malignant (cancerous), in which case it is known as eye cancer. Eye cancers can be primary or metastatic cancer. The two most common cancers that spread to the eye from another organ are breast cancer and lung cancer. Other less common sites of origin include the prostate, kidney, thyroid, skin, colon and blood or bone marrow.
Optic neuropathy is damage to the optic nerve from any cause. The optic nerve is a bundle of millions of fibers in the retina that sends visual signals to the brain.
Intraocular lymphoma is a rare malignant form of eye cancer. Intraocular lymphoma may affect the eye secondarily from a metastasis from a non-ocular tumor or may arise within the eye primarily. PIOL is a subset of primary central nervous system lymphoma (PCNSL). PCNSL are most commonly a diffuse large B-cell immunohistologic subtype of non-Hodgkin's lymphoma according to the World Health Organization (WHO) classification of lymphomas. The most common symptoms of PIOL include blurred or decreased vision due to tumor cells in the vitreous. Most cases of PIOL eventuate to central nervous system involvement (PCNSL) while only 20% of PCNSL lead to intraocular (PIOL) involvement. PIOL and PCNSL remain enigmas because both structures are immunologically privileged sites and so do not normally have immune cells trafficking through these structures. What is more, while the vast majority of PCNSL in patients with acquired immune deficiency syndrome (AIDS) is related to the Epstein-Barr virus (EBV), the development of PCNSL and PIOL in immunocompetent patients is unknown and shows no general relation to infectious DNAs.
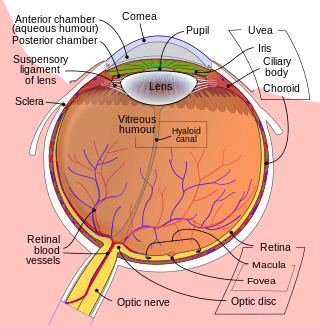
Intraocular hemorrhage is bleeding inside the eye. Bleeding can occur from any structure of the eye where there is vasculature or blood flow, including the anterior chamber, vitreous cavity, retina, choroid, suprachoroidal space, or optic disc.

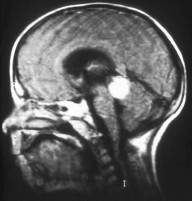
Pineoblastoma is a malignant tumor of the pineal gland. A pineoblastoma is a supratentorial midline primitive neuroectodermal tumor. Pineoblastoma can present at any age, but is most common in young children. They account for 0.001% of all primary CNS neoplasms.

Persistent fetal vasculature(PFV), also known as persistent fetal vasculature syndrome (PFVS), and until 1997 known primarily as persistent hyperplastic primary vitreous (PHPV), is a rare congenital anomaly which occurs when blood vessels within the developing eye, known as the embryonic hyaloid vasculature network, fail to regress as they normally would in-utero after the eye is fully developed. Defects which arise from this lack of vascular regression are diverse; as a result, the presentation, symptoms, and prognosis of affected patients vary widely, ranging from clinical insignificance to irreversible blindness. The underlying structural causes of PFV are considered to be relatively common, and the vast majority of cases do not warrant additional intervention. When symptoms do manifest, however, they are often significant, causing detrimental and irreversible visual impairment. Persistent fetal vasculature heightens the lifelong risk of glaucoma, cataracts, intraocular hemorrhages, and Retinal detachments, accounting for the visual loss of nearly 5% of the blind community in the developed world. In diagnosed cases of PFV, approximately 90% of patients with a unilateral disease have associated poor vision in the affected eye.
Retinal gene therapy holds a promise in treating different forms of non-inherited and inherited blindness.

Orbital lymphoma is a common type of non-Hodgkin lymphoma that occurs near or on the eye. Common symptoms include decreased vision and uveitis. Orbital lymphoma can be diagnosed via a biopsy of the eye and is usually treated with radiotherapy or in combination with chemotherapy.
Targeted molecular therapy for neuroblastoma involves treatment aimed at molecular targets that have a unique expression in this form of cancer. Neuroblastoma, the second most common pediatric malignant tumor, often involves treatment through intensive chemotherapy. A number of molecular targets have been identified for the treatment of high-risk forms of this disease. Aiming treatment in this way provides a more selective way to treat the disease, decreasing the risk for toxicities that are associated with the typical treatment regimen. Treatment using these targets can supplement or replace some of the intensive chemotherapy that is used for neuroblastoma. These molecular targets of this disease include GD2, ALK, and CD133. GD2 is a target of immunotherapy, and is the most fully developed of these treatment methods, but is also associated with toxicities. ALK has more recently been discovered, and drugs in development for this target are proving to be successful in neuroblastoma treatment. The role of CD133 in neuroblastoma has also been more recently discovered and is an effective target for treatment of this disease.

J. William Harbour is an American ophthalmologist, ocular oncologist and cancer researcher. He is Chair of the Department of Ophthalmology at the University of Texas Southwestern Medical Center in Dallas. He previously served as the vice chair and director of ocular oncology at the Bascom Palmer Eye Institute and associate director for basic science at the Sylvester Comprehensive Cancer Center of the University of Miami's Miller School of Medicine.
Occult macular dystrophy (OMD) is a rare inherited degradation of the retina, characterized by progressive loss of function in the most sensitive part of the central retina (macula), the location of the highest concentration of light-sensitive cells (photoreceptors) but presenting no visible abnormality. "Occult" refers to the degradation in the fundus being difficult to discern. The disorder is called "dystrophy" instead of "degradation" to distinguish its genetic origin from other causes, such as age. OMD was first reported by Y. Miyake et al. in 1989.

Congenital blindness refers to blindness present at birth. Congenital blindness is sometimes used interchangeably with "Childhood Blindness." However, current literature has various definitions of both terms. Childhood blindness encompasses multiple diseases and conditions present in ages up to 16 years old, which can result in permanent blindness or severe visual impairment over time. Congenital blindness is a hereditary disease and can be treated by gene therapy. Visual loss in children or infants can occur either at the prenatal stage or postnatal stage. There are multiple possible causes of congenital blindness. In general, 60% of congenital blindness cases are contributed from prenatal stage and 40% are contributed from inherited disease. However, most of the congenital blindness cases show that it can be avoidable or preventable with early treatment.
Sickle cell retinopathy can be defined as retinal changes due to blood vessel damage in the eye of a person with a background of sickle cell disease. It can likely progress to loss of vision in late stages due to vitreous hemorrhage or retinal detachment. Sickle cell disease is a structural red blood cell disorder leading to consequences in multiple systems. It is characterized by chronic red blood cell destruction, vascular injury, and tissue ischemia causing damage to the brain, eyes, heart, lungs, kidneys, spleen, and musculoskeletal system.