Related Research Articles
In bioinformatics, sequence clustering algorithms attempt to group biological sequences that are somehow related. The sequences can be either of genomic, "transcriptomic" (ESTs) or protein origin. For proteins, homologous sequences are typically grouped into families. For EST data, clustering is important to group sequences originating from the same gene before the ESTs are assembled to reconstruct the original mRNA.

A protein family is a group of evolutionarily related proteins. In many cases, a protein family has a corresponding gene family, in which each gene encodes a corresponding protein with a 1:1 relationship. The term "protein family" should not be confused with family as it is used in taxonomy.
In genetics, an expressed sequence tag (EST) is a short sub-sequence of a cDNA sequence. ESTs may be used to identify gene transcripts, and were instrumental in gene discovery and in gene-sequence determination. The identification of ESTs has proceeded rapidly, with approximately 74.2 million ESTs now available in public databases. EST approaches have largely been superseded by whole genome and transcriptome sequencing and metagenome sequencing.
In computational biology, gene prediction or gene finding refers to the process of identifying the regions of genomic DNA that encode genes. This includes protein-coding genes as well as RNA genes, but may also include prediction of other functional elements such as regulatory regions. Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced.
The transcriptome is the set of all RNA transcripts, including coding and non-coding, in an individual or a population of cells. The term can also sometimes be used to refer to all RNAs, or just mRNA, depending on the particular experiment. The term transcriptome is a portmanteau of the words transcript and genome; it is associated with the process of transcript production during the biological process of transcription.
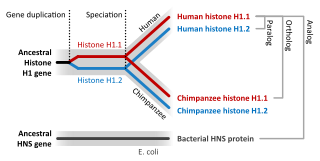
Sequence homology is the biological homology between DNA, RNA, or protein sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry because of three phenomena: either a speciation event (orthologs), or a duplication event (paralogs), or else a horizontal gene transfer event (xenologs).

MicrobesOnline is a publicly and freely accessible website that hosts multiple comparative genomic tools for comparing microbial species at the genomic, transcriptomic and functional levels. MicrobesOnline was developed by the Virtual Institute for Microbial Stress and Survival, which is based at the Lawrence Berkeley National Laboratory in Berkeley, California. The site was launched in 2005, with regular updates until 2011.

RNA-Seq is a sequencing technique which uses next-generation sequencing (NGS) to reveal the presence and quantity of RNA in a biological sample at a given moment, analyzing the continuously changing cellular transcriptome.
GeneCards is a database of human genes that provides genomic, proteomic, transcriptomic, genetic and functional information on all known and predicted human genes. It is being developed and maintained by the Crown Human Genome Center at the Weizmann Institute of Science, in collaboration with LifeMap Sciences.
OrthoDB presents a catalog of orthologous protein-coding genes across vertebrates, arthropods, fungi, plants, and bacteria. Orthology refers to the last common ancestor of the species under consideration, and thus OrthoDB explicitly delineates orthologs at each major radiation along the species phylogeny. The database of orthologs presents available protein descriptors, together with Gene Ontology and InterPro attributes, which serve to provide general descriptive annotations of the orthologous groups, and facilitate comprehensive orthology database querying. OrthoDB also provides computed evolutionary traits of orthologs, such as gene duplicability and loss profiles, divergence rates, sibling groups, and gene intron-exon architectures.
PhylomeDB is a public biological database for complete catalogs of gene phylogenies (phylomes). It allows users to interactively explore the evolutionary history of genes through the visualization of phylogenetic trees and multiple sequence alignments. Moreover, phylomeDB provides genome-wide orthology and paralogy predictions which are based on the analysis of the phylogenetic trees. The automated pipeline used to reconstruct trees aims at providing a high-quality phylogenetic analysis of different genomes, including Maximum Likelihood tree inference, alignment trimming and evolutionary model testing.
De novo transcriptome assembly is the de novo sequence assembly method of creating a transcriptome without the aid of a reference genome.
Chimeric RNA, sometimes referred to as a fusion transcript, is composed of exons from two or more different genes that have the potential to encode novel proteins. These mRNAs are different from those produced by conventional splicing as they are produced by two or more gene loci.
Metatranscriptomics is the science that studies gene expression of microbes within natural environments, i.e., the metatranscriptome. It also allows to obtain whole gene expression profiling of complex microbial communities.
TopHat is an open-source bioinformatics tool for the throughput alignment of shotgun cDNA sequencing reads generated by transcriptomics technologies using Bowtie first and then mapping to a reference genome to discover RNA splice sites de novo. TopHat aligns RNA-Seq reads to mammalian-sized genomes.
Single-cell transcriptomics examines the gene expression level of individual cells in a given population by simultaneously measuring the RNA concentration of hundreds to thousands of genes. Single-cell transcriptomics makes it possible to unravel heterogeneous cell populations, reconstruct cellular developmental pathways, and model transcriptional dynamics — all previously masked in bulk RNA sequencing.
Transcriptomics technologies are the techniques used to study an organism's transcriptome, the sum of all of its RNA transcripts. The information content of an organism is recorded in the DNA of its genome and expressed through transcription. Here, mRNA serves as a transient intermediary molecule in the information network, whilst non-coding RNAs perform additional diverse functions. A transcriptome captures a snapshot in time of the total transcripts present in a cell. Transcriptomics technologies provide a broad account of which cellular processes are active and which are dormant. A major challenge in molecular biology is to understand how a single genome gives rise to a variety of cells. Another is how gene expression is regulated.

OrthoFinder is a command-line software tool for comparative genomics. OrthoFinder determines the correspondence between genes in different organisms. This correspondence provides a framework for understanding the evolution of life on Earth, and enables the extrapolation and transfer of biological knowledge between organisms.
References
- 1 2 3 Hörandl, Elvira; Appelhans, Mark (2015). Next-generation sequencing in plant systematics. Koeltz Scientific Books. ISBN 9783874294928.
- 1 2 Salichos, Leonidas; Rokas, Antonis; Fairhead, Cecile (13 April 2011). "Evaluating Ortholog Prediction Algorithms in a Yeast Model Clade". PLOS ONE. 6 (4): e18755. doi: 10.1371/journal.pone.0018755 . PMC 3076445 . PMID 21533202.
- ↑ Ostlund, G.; Schmitt, T.; Forslund, K.; Kostler, T.; Messina, D. N.; Roopra, S.; Frings, O.; Sonnhammer, E. L. L. (5 November 2009). "InParanoid 7: new algorithms and tools for eukaryotic orthology analysis". Nucleic Acids Research. 38 (Database): D196–D203. doi:10.1093/nar/gkp931. PMC 2808972 . PMID 19892828.
- ↑ Alexeyenko, A.; Tamas, I.; Liu, G.; Sonnhammer, E. L.L. (27 July 2006). "Automatic clustering of orthologs and inparalogs shared by multiple proteomes". Bioinformatics. 22 (14): e9–e15. doi: 10.1093/bioinformatics/btl213 . PMID 16873526.
- ↑ Li, L. (1 September 2003). "OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes". Genome Research. 13 (9): 2178–2189. doi:10.1101/gr.1224503. PMC 403725 . PMID 12952885.
- ↑ Sayers, E. W.; Barrett, T.; Benson, D. A.; Bolton, E.; Bryant, S. H.; Canese, K.; Chetvernin, V.; Church, D. M.; DiCuccio, M.; Federhen, S.; Feolo, M.; Fingerman, I. M.; Geer, L. Y.; Helmberg, W.; Kapustin, Y.; Landsman, D.; Lipman, D. J.; Lu, Z.; Madden, T. L.; Madej, T.; Maglott, D. R.; Marchler-Bauer, A.; Miller, V.; Mizrachi, I.; Ostell, J.; Panchenko, A.; Phan, L.; Pruitt, K. D.; Schuler, G. D.; Sequeira, E.; Sherry, S. T.; Shumway, M.; Sirotkin, K.; Slotta, D.; Souvorov, A.; Starchenko, G.; Tatusova, T. A.; Wagner, L.; Wang, Y.; Wilbur, W. J.; Yaschenko, E.; Ye, J. (21 November 2010). "Database resources of the National Center for Biotechnology Information". Nucleic Acids Research. 39 (Database): D38–D51. doi:10.1093/nar/gkq1172. PMC 3013733 . PMID 21097890.
- ↑ Altenhoff, A. M.; kunca, N.; Glover, N.; Train, C.-M.; Sueki, A.; Pili ota, I.; Gori, K.; Tomiczek, B.; Muller, S.; Redestig, H.; Gonnet, G. H.; Dessimoz, C. (15 November 2014). "The OMA orthology database in 2015: function predictions, better plant support, synteny view and other improvements". Nucleic Acids Research. 43 (D1): D240–D249. doi: 10.1093/nar/gku1158 . PMC 4383958 . PMID 25399418.
- ↑ Zmasek, Christian M; Eddy, Sean R (2002). "RIO: Analyzing proteomes by automated phylogenomics using resampled inference of orthologs". BMC Bioinformatics. 3 (1): 14. doi: 10.1186/1471-2105-3-14 . PMC 116988 . PMID 12028595.
- ↑ Barker, M. S.; Vogel, H.; Schranz, M. E. (5 October 2009). "Paleopolyploidy in the Brassicales: Analyses of the Cleome Transcriptome Elucidate the History of Genome Duplications in Arabidopsis and Other Brassicales". Genome Biology and Evolution. 1: 391–399. doi: 10.1093/gbe/evp040 . PMC 2817432 . PMID 20333207.
- ↑ Yang, Xu; Cheng, Yu-Fu; Deng, Cao; Ma, Yan; Wang, Zhi-Wen; Chen, Xue-Hao; Xue, Lin-Bao (2014). "Comparative transcriptome analysis of eggplant (Solanum melongena L.) and turkey berry (Solanum torvum Sw.): phylogenomics and disease resistance analysis". BMC Genomics. 15 (1): 412. doi: 10.1186/1471-2164-15-412 . PMC 4070557 . PMID 24885385.
- ↑ Wernersson, R. (1 July 2003). "RevTrans: multiple alignment of coding DNA from aligned amino acid sequences". Nucleic Acids Research. 31 (13): 3537–3539. doi: 10.1093/nar/gkg609 . PMC 169015 . PMID 12824361.
- ↑ Moreno-Hagelsieb, G.; Latimer, K. (26 November 2007). "Choosing BLAST options for better detection of orthologs as reciprocal best hits". Bioinformatics. 24 (3): 319–324. doi: 10.1093/bioinformatics/btm585 . PMID 18042555.
- ↑ Castillo-Ramírez, Santiago; González, Víctor (2008). "Factors affecting the concordance between orthologous gene trees and species tree in bacteria". BMC Evolutionary Biology. 8 (1): 300. doi: 10.1186/1471-2148-8-300 . PMC 2614993 . PMID 18973688.
- ↑ Wen, Jun; Xiong, Zhiqiang; Nie, Ze-Long; Mao, Likai; Zhu, Yabing; Kan, Xian-Zhao; Ickert-Bond, Stefanie M.; Gerrath, Jean; Zimmer, Elizabeth A.; Fang, Xiao-Dong; Candela, Hector (17 September 2013). "Transcriptome Sequences Resolve Deep Relationships of the Grape Family". PLOS ONE. 8 (9): e74394. doi: 10.1371/journal.pone.0074394 . PMC 3775763 . PMID 24069307.