
Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.
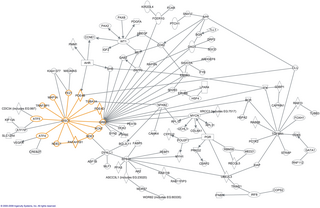
In molecular biology, an interactome is the whole set of molecular interactions in a particular cell. The term specifically refers to physical interactions among molecules but can also describe sets of indirect interactions among genes.
A biochemical cascade, also known as a signaling cascade or signaling pathway, is a series of chemical reactions that occur within a biological cell when initiated by a stimulus. This stimulus, known as a first messenger, acts on a receptor that is transduced to the cell interior through second messengers which amplify the signal and transfer it to effector molecules, causing the cell to respond to the initial stimulus. Most biochemical cascades are series of events, in which one event triggers the next, in a linear fashion. At each step of the signaling cascade, various controlling factors are involved to regulate cellular actions, in order to respond effectively to cues about their changing internal and external environments.
The European Bioinformatics Institute (EMBL-EBI) is an intergovernmental organization (IGO) which, as part of the European Molecular Biology Laboratory (EMBL) family, focuses on research and services in bioinformatics. It is located on the Wellcome Genome Campus in Hinxton near Cambridge, and employs over 600 full-time equivalent (FTE) staff. Institute leaders such as Rolf Apweiler, Alex Bateman, Ewan Birney, and Guy Cochrane, an adviser on the National Genomics Data Center Scientific Advisory Board, serve as part of the international research network of the BIG Data Center at the Beijing Institute of Genomics.

Protein–protein interactions (PPIs) are physical contacts of high specificity established between two or more protein molecules as a result of biochemical events steered by interactions that include electrostatic forces, hydrogen bonding and the hydrophobic effect. Many are physical contacts with molecular associations between chains that occur in a cell or in a living organism in a specific biomolecular context.

KEGG is a collection of databases dealing with genomes, biological pathways, diseases, drugs, and chemical substances. KEGG is utilized for bioinformatics research and education, including data analysis in genomics, metagenomics, metabolomics and other omics studies, modeling and simulation in systems biology, and translational research in drug development.

Dame Janet Maureen Thornton, is a senior scientist and director emeritus at the European Bioinformatics Institute (EBI), part of the European Molecular Biology Laboratory (EMBL). She is one of the world's leading researchers in structural bioinformatics, using computational methods to understand protein structure and function. She served as director of the EBI from October 2001 to June 2015, and played a key role in ELIXIR.
BioPAX is a RDF/OWL-based standard language to represent biological pathways at the molecular and cellular level. Its major use is to facilitate the exchange of pathway data. Pathway data captures our understanding of biological processes, but its rapid growth necessitates development of databases and computational tools to aid interpretation. However, the current fragmentation of pathway information across many databases with incompatible formats presents barriers to its effective use. BioPAX solves this problem by making pathway data substantially easier to collect, index, interpret and share. BioPAX can represent metabolic and signaling pathways, molecular and genetic interactions and gene regulation networks. BioPAX was created through a community process. Through BioPAX, millions of interactions organized into thousands of pathways across many organisms, from a growing number of sources, are available. Thus, large amounts of pathway data are available in a computable form to support visualization, analysis and biological discovery.

In molecular biology, STRING is a biological database and web resource of known and predicted protein–protein interactions.
The ConsensusPathDB is a molecular functional interaction database, integrating information on protein interactions, genetic interactions signaling, metabolism, gene regulation, and drug-target interactions in humans. ConsensusPathDB currently includes such interactions from 32 databases. ConsensusPathDB is freely available for academic use under http://ConsensusPathDB.org.
A biological network is a method of representing systems as complex sets of binary interactions or relations between various biological entities. In general, networks or graphs are used to capture relationships between entities or objects. A typical graphing representation consists of a set of nodes connected by edges.
GeneCards is a database of human genes that provides genomic, proteomic, transcriptomic, genetic and functional information on all known and predicted human genes. It is being developed and maintained by the Crown Human Genome Center at the Weizmann Institute of Science, in collaboration with LifeMap Sciences.
A biological pathway is a series of interactions among molecules in a cell that leads to a certain product or a change in a cell. Such a pathway can trigger the assembly of new molecules, such as a fat or protein. Pathways can also turn genes on and off, or spur a cell to move. Some of the most common biological pathways are involved in metabolism, the regulation of gene expression and the transmission of signals. Pathways play a key role in advanced studies of genomics.

WikiPathways is a community resource for contributing and maintaining content dedicated to biological pathways. Any registered WikiPathways user can contribute, and anybody can become a registered user. Contributions are monitored by a group of admins, but the bulk of peer review, editorial curation, and maintenance is the responsibility of the user community. WikiPathways is originally built using MediaWiki software, a custom graphical pathway editing tool (PathVisio) and integrated BridgeDb databases covering major gene, protein, and metabolite systems. WikiPathways was founded in 2008 by Thomas Kelder, Alex Pico, Martijn Van Iersel, Kristina Hanspers, Bruce Conklin and Chris Evelo. Current architects are Alex Pico and Martina Summer-Kutmon.
Protein function prediction methods are techniques that bioinformatics researchers use to assign biological or biochemical roles to proteins. These proteins are usually ones that are poorly studied or predicted based on genomic sequence data. These predictions are often driven by data-intensive computational procedures. Information may come from nucleic acid sequence homology, gene expression profiles, protein domain structures, text mining of publications, phylogenetic profiles, phenotypic profiles, and protein-protein interaction. Protein function is a broad term: the roles of proteins range from catalysis of biochemical reactions to transport to signal transduction, and a single protein may play a role in multiple processes or cellular pathways.

Lincoln David Stein is a scientist and Professor in bioinformatics and computational biology at the Ontario Institute for Cancer Research.
Virtual Cell (VCell) is an open-source software platform for modeling and simulation of living organisms, primarily cells. It has been designed to be a tool for a wide range of scientists, from experimental cell biologists to theoretical biophysicists.
In bioinformatics, the PANTHER classification system is a large curated biological database of gene/protein families and their functionally related subfamilies that can be used to classify and identify the function of gene products. PANTHER is part of the Gene Ontology Reference Genome Project designed to classify proteins and their genes for high-throughput analysis.

Pathway is the term from molecular biology for a curated schematic representation of a well characterized segment of the molecular physiological machinery, such as a metabolic pathway describing an enzymatic process within a cell or tissue or a signaling pathway model representing a regulatory process that might, in its turn, enable a metabolic or another regulatory process downstream. A typical pathway model starts with an extracellular signaling molecule that activates a specific receptor, thus triggering a chain of molecular interactions. A pathway is most often represented as a relatively small graph with gene, protein, and/or small molecule nodes connected by edges of known functional relations. While a simpler pathway might appear as a chain, complex pathway topologies with loops and alternative routes are much more common. Computational analyses employ special formats of pathway representation. In the simplest form, however, a pathway might be represented as a list of member molecules with order and relations unspecified. Such a representation, generally called Functional Gene Set (FGS), can also refer to other functionally characterised groups such as protein families, Gene Ontology (GO) and Disease Ontology (DO) terms etc. In bioinformatics, methods of pathway analysis might be used to identify key genes/ proteins within a previously known pathway in relation to a particular experiment / pathological condition or building a pathway de novo from proteins that have been identified as key affected elements. By examining changes in e.g. gene expression in a pathway, its biological activity can be explored. However most frequently, pathway analysis refers to a method of initial characterization and interpretation of an experimental condition that was studied with omics tools or genome-wide association study. Such studies might identify long lists of altered genes. A visual inspection is then challenging and the information is hard to summarize, since the altered genes map to a broad range of pathways, processes, and molecular functions. In such situations, the most productive way of exploring the list is to identify enrichment of specific FGSs in it. The general approach of enrichment analyses is to identify FGSs, members of which were most frequently or most strongly altered in the given condition, in comparison to a gene set sampled by chance. In other words, enrichment can map canonical prior knowledge structured in the form of FGSs to the condition represented by altered genes.