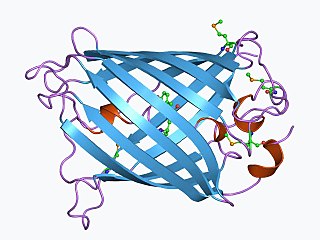
The green fluorescent protein (GFP) is a protein that exhibits bright green fluorescence when exposed to light in the blue to ultraviolet range. The label GFP traditionally refers to the protein first isolated from the jellyfish Aequorea victoria and is sometimes called avGFP. However, GFPs have been found in other organisms including corals, sea anemones, zoanithids, copepods and lancelets.

In molecular biology and biotechnology, a fluorescent tag, also known as a fluorescent label or fluorescent probe, is a molecule that is attached chemically to aid in the detection of a biomolecule such as a protein, antibody, or amino acid. Generally, fluorescent tagging, or labeling, uses a reactive derivative of a fluorescent molecule known as a fluorophore. The fluorophore selectively binds to a specific region or functional group on the target molecule and can be attached chemically or biologically. Various labeling techniques such as enzymatic labeling, protein labeling, and genetic labeling are widely utilized. Ethidium bromide, fluorescein and green fluorescent protein are common tags. The most commonly labelled molecules are antibodies, proteins, amino acids and peptides which are then used as specific probes for detection of a particular target.
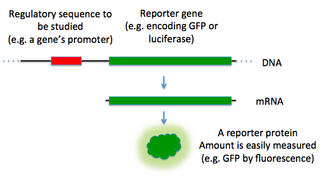
In molecular biology, a reporter gene is a gene that researchers attach to a regulatory sequence of another gene of interest in bacteria, cell culture, animals or plants. Such genes are called reporters because the characteristics they confer on organisms expressing them are easily identified and measured, or because they are selectable markers. Reporter genes are often used as an indication of whether a certain gene has been taken up by or expressed in the cell or organism population.
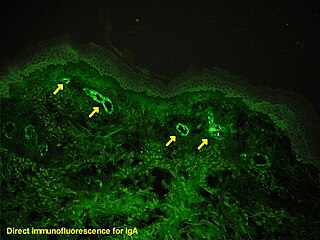
Immunofluorescence is a technique used for light microscopy with a fluorescence microscope and is used primarily on microbiological samples. This technique uses the specificity of antibodies to their antigen to target fluorescent dyes to specific biomolecule targets within a cell, and therefore allows visualization of the distribution of the target molecule through the sample. The specific region an antibody recognizes on an antigen is called an epitope. There have been efforts in epitope mapping since many antibodies can bind the same epitope and levels of binding between antibodies that recognize the same epitope can vary. Additionally, the binding of the fluorophore to the antibody itself cannot interfere with the immunological specificity of the antibody or the binding capacity of its antigen. Immunofluorescence is a widely used example of immunostaining and is a specific example of immunohistochemistry. This technique primarily makes use of fluorophores to visualise the location of the antibodies.

Fluorescence recovery after photobleaching (FRAP) is a method for determining the kinetics of diffusion through tissue or cells. It is capable of quantifying the two dimensional lateral diffusion of a molecularly thin film containing fluorescently labeled probes, or to examine single cells. This technique is very useful in biological studies of cell membrane diffusion and protein binding. In addition, surface deposition of a fluorescing phospholipid bilayer allows the characterization of hydrophilic surfaces in terms of surface structure and free energy.

Förster or fluorescence resonance energy transfer (FRET), resonance energy transfer (RET) or electronic energy transfer (EET) is a mechanism describing energy transfer between two light-sensitive molecules (chromophores). A donor chromophore, initially in its electronic excited state, may transfer energy to an acceptor chromophore through nonradiative dipole–dipole coupling. The efficiency of this energy transfer is inversely proportional to the sixth power of the distance between donor and acceptor, making FRET extremely sensitive to small changes in distance.

A fluorescence microscope is an optical microscope that uses fluorescence instead of, or in addition to, scattering, reflection, and attenuation or absorption, to study the properties of organic or inorganic substances. "Fluorescence microscope" refers to any microscope that uses fluorescence to generate an image, whether it is a simple set up like an epifluorescence microscope or a more complicated design such as a confocal microscope, which uses optical sectioning to get better resolution of the fluorescence image.

Two-hybrid screening is a molecular biology technique used to discover protein–protein interactions (PPIs) and protein–DNA interactions by testing for physical interactions between two proteins or a single protein and a DNA molecule, respectively.
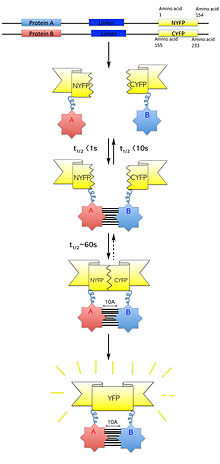
Within the field of molecular biology, a protein-fragment complementation assay, or PCA, is a method for the identification and quantification of protein–protein interactions. In the PCA, the proteins of interest are each covalently linked to fragments of a third protein. Interaction between the bait and the prey proteins brings the fragments of the reporter protein in close proximity to allow them to form a functional reporter protein whose activity can be measured. This principle can be applied to many different reporter proteins and is also the basis for the yeast two-hybrid system, an archetypical PCA assay.
The GFP-cDNA project documents the localisation of proteins to subcellular compartments of the eukaryotic cell applying fluorescence microscopy. Experimental data are complemented with bioinformatic analyses and published online in a database. A search function allows the finding of proteins containing features or motifs of particular interest. The project is a collaboration of the research groups of Rainer Pepperkok at the European Molecular Biology Laboratory (EMBL) and Stefan Wiemann at the German Cancer Research Centre (DKFZ).

Fusion proteins or chimeric (kī-ˈmir-ik) proteins are proteins created through the joining of two or more genes that originally coded for separate proteins. Translation of this fusion gene results in a single or multiple polypeptides with functional properties derived from each of the original proteins. Recombinant fusion proteins are created artificially by recombinant DNA technology for use in biological research or therapeutics. Chimeric or chimera usually designate hybrid proteins made of polypeptides having different functions or physico-chemical patterns. Chimeric mutant proteins occur naturally when a complex mutation, such as a chromosomal translocation, tandem duplication, or retrotransposition creates a novel coding sequence containing parts of the coding sequences from two different genes. Naturally occurring fusion proteins are commonly found in cancer cells, where they may function as oncoproteins. The bcr-abl fusion protein is a well-known example of an oncogenic fusion protein, and is considered to be the primary oncogenic driver of chronic myelogenous leukemia.
DNA adenine methyltransferase identification, often abbreviated DamID, is a molecular biology protocol used to map the binding sites of DNA- and chromatin-binding proteins in eukaryotes. DamID identifies binding sites by expressing the proposed DNA-binding protein as a fusion protein with DNA methyltransferase. Binding of the protein of interest to DNA localizes the methyltransferase in the region of the binding site. Adenine methylation does not occur naturally in eukaryotes and therefore adenine methylation in any region can be concluded to have been caused by the fusion protein, implying the region is located near a binding site. DamID is an alternate method to ChIP-on-chip or ChIP-seq.
EosFP is a photoactivatable green to red fluorescent protein. Its green fluorescence (516 nm) switches to red (581 nm) upon UV irradiation of ~390 nm due to a photo-induced modification resulting from a break in the peptide backbone near the chromophore. Eos was first discovered as a tetrameric protein in the stony coral Lobophyllia hemprichii. Like other fluorescent proteins, Eos allows for applications such as the tracking of fusion proteins, multicolour labelling and tracking of cell movement. Several variants of Eos have been engineered for use in specific study systems including mEos2, mEos4 and CaMPARI.

Fluorescence is used in the life sciences generally as a non-destructive way of tracking or analysing biological molecules. Some proteins or small molecules in cells are naturally fluorescent, which is called intrinsic fluorescence or autofluorescence. Alternatively, specific or general proteins, nucleic acids, lipids or small molecules can be "labelled" with an extrinsic fluorophore, a fluorescent dye which can be a small molecule, protein or quantum dot. Several techniques exist to exploit additional properties of fluorophores, such as fluorescence resonance energy transfer, where the energy is passed non-radiatively to a particular neighbouring dye, allowing proximity or protein activation to be detected; another is the change in properties, such as intensity, of certain dyes depending on their environment allowing their use in structural studies.
MS2 tagging is a technique based upon the natural interaction of the MS2 bacteriophage coat protein with a stem-loop structure from the phage genome, which is used for biochemical purification of RNA-protein complexes and partnered to GFP for detection of RNA in living cells. More recently, the technique has been used to monitor the appearance of RNA in living cells, at the site of transcription, or simply by observing the changes in RNA number in the cytoplasm. This has revealed that transcription of both prokaryotic and eukaryotic genes occurs in a discontinuous fashion with bursts of transcription separated by irregular intervals.
There are many methods to investigate protein–protein interactions which are the physical contacts of high specificity established between two or more protein molecules involving electrostatic forces and hydrophobic effects. Each of the approaches has its own strengths and weaknesses, especially with regard to the sensitivity and specificity of the method. A high sensitivity means that many of the interactions that occur are detected by the screen. A high specificity indicates that most of the interactions detected by the screen are occurring in reality.

SNAP-tag® is a self-labeling protein tag commercially available in various expression vectors. SNAP-tag is a 182 residues polypeptide that can be fused to any protein of interest and further specifically and covalently tagged with a suitable ligand, such as a fluorescent dye. Since its introduction, SNAP-tag has found numerous applications in biochemistry and for the investigation of the function and localisation of proteins and enzymes in living cells. Compared to the current standard labelling methods used in fluorescence microscopy, the use of SNAP-tag presents significant advantages. SNAP-tag® is a registered trademark of New England Biolabs, Inc.
PRIME is a molecular biology research tool developed by Alice Y. Ting and the Ting Lab at MIT for site-specific labeling of proteins in living cells with chemical probes. Probes often have useful biophysical properties, such as fluorescence, and allow imaging of proteins. Ultimately, PRIME enables scientists to study functions of specific proteins of interest.

The need for fluorescently tracking RNA rose as its roles in complex cellular functions has grown to not only include mRNA, rRNA, and tRNA, but also RNAi, siRNA, snoRNA, and lncRNA, among others. Spinach is a synthetically derived RNA aptamer born out of the need for a way of studying the role of RNAs at the cellular level. This aptamer was created using Systematic Evolution for Ligands by EXponential enrichment, or SELEX, which is also known as in vitro evolution.
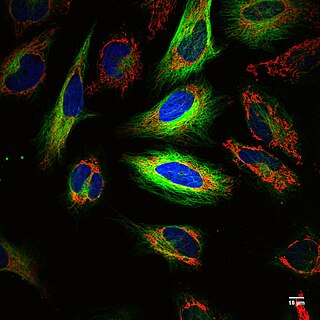
Fluorescence imaging is a type of non-invasive imaging technique that can help visualize biological processes taking place in a living organism. Images can be produced from a variety of methods including: microscopy, imaging probes, and spectroscopy.