Kocher–Debré–Semelaigne syndrome(KDSS) is hypothyroidism in infancy or childhood characterised by lower extremity or generalized muscular hypertrophy (Herculean appearance), myxoedema, short stature, and cognitive impairment.
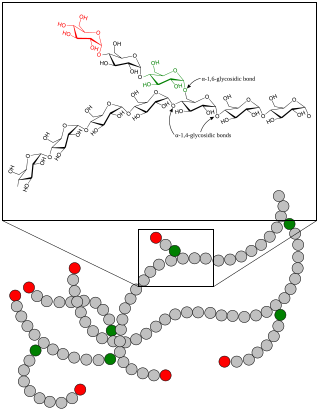
A glycogen storage disease is a metabolic disorder caused by a deficiency of an enzyme or transport protein affecting glycogen synthesis, glycogen breakdown, or glucose breakdown, typically in muscles and/or liver cells.

Skeletal muscles are organs of the vertebrate muscular system and typically are attached by tendons to bones of a skeleton. The muscle cells of skeletal muscles are much longer than in the other types of muscle tissue, and are often known as muscle fibers. The muscle tissue of a skeletal muscle is striated – having a striped appearance due to the arrangement of the sarcomeres.
Myotonia is a symptom of a small handful of certain neuromuscular disorders characterized by delayed relaxation of the skeletal muscles after voluntary contraction or electrical stimulation, and the muscle shows an abnormal EMG.

Dantrolene sodium, sold under the brand name Dantrium among others, is a postsynaptic muscle relaxant that lessens excitation-contraction coupling in muscle cells. It achieves this by inhibiting Ca2+ ions release from sarcoplasmic reticulum stores by antagonizing ryanodine receptors. It is the primary drug used for the treatment and prevention of malignant hyperthermia, a rare, life-threatening disorder triggered by general anesthesia or drugs. It is also used in the management of neuroleptic malignant syndrome, muscle spasticity (e.g. after strokes, in paraplegia, cerebral palsy, or patients with multiple sclerosis), and poisoning by 2,4-dinitrophenol or by the related compounds dinoseb and dinoterb.
Muscle fatigue is when muscles that were initially generating a normal amount of force, then experience a declining ability to generate force. It can be a result of vigorous exercise, but abnormal fatigue may be caused by barriers to or interference with the different stages of muscle contraction. There are two main causes of muscle fatigue: the limitations of a nerve’s ability to generate a sustained signal ; and the reduced ability of the muscle fiber to contract.
Hyperkalemic periodic paralysis is an inherited autosomal dominant disorder that affects sodium channels in muscle cells and the ability to regulate potassium levels in the blood. It is characterized by muscle hyperexcitability or weakness which, exacerbated by potassium, heat or cold, can lead to uncontrolled shaking followed by paralysis. Onset usually occurs in early childhood, but it still occurs with adults.
Nemaline myopathy is a congenital, often hereditary neuromuscular disorder with many symptoms that can occur such as muscle weakness, hypoventilation, swallowing dysfunction, and impaired speech ability. The severity of these symptoms varies and can change throughout one's life to some extent. The prevalence is estimated at 1 in 50,000 live births. It is the most common non-dystrophic myopathy.

Channelopathies are a group of diseases caused by the dysfunction of ion channel subunits or their interacting proteins. These diseases can be inherited or acquired by other disorders, drugs, or toxins. Mutations in genes encoding ion channels, which impair channel function, are the most common cause of channelopathies. There are more than 400 genes that encode ion channels, found in all human cell types and are involved in almost all physiological processes. Each type of channel is a multimeric complex of subunits encoded by a number of genes. Depending where the mutation occurs it may affect the gating, conductance, ion selectivity, or signal transduction of the channel.
Myotonia congenita is a congenital neuromuscular channelopathy that affects skeletal muscles. It is a genetic disorder. The hallmark of the disease is the failure of initiated contraction to terminate, often referred to as delayed relaxation of the muscles (myotonia) and rigidity. Symptoms include delayed relaxation of the muscles after voluntary contraction (myotonia), and may also include stiffness, hypertrophy (enlargement), transient weakness in some forms of the disorder, severe masseter spasm, and cramping. The condition is sometimes referred to as fainting goat syndrome, as it is responsible for the eponymous 'fainting' seen in fainting goats when presented with a sudden stimulus. Of note, myotonia congenita has no association with malignant hyperthermia (MH).

Mitochondrial myopathies are types of myopathies associated with mitochondrial disease. Adenosine triphosphate (ATP), the chemical used to provide energy for the cell, cannot be produced sufficiently by oxidative phosphorylation when the mitochondrion is either damaged or missing necessary enzymes or transport proteins. With ATP production deficient in mitochondria, there is an over-reliance on anaerobic glycolysis which leads to lactic acidosis either at rest or exercise-induced.
Periodic paralysis is a group of rare genetic diseases that lead to weakness or paralysis from common triggers such as cold, heat, high carbohydrate meals, not eating, stress or excitement and physical activity of any kind. The underlying mechanism of these diseases are malfunctions in the ion channels in skeletal muscle cell membranes that allow electrically charged ions to leak in or out of the muscle cell, causing the cell to depolarize and become unable to move.

Paramyotonia congenita (PC) is a rare congenital autosomal dominant neuromuscular disorder characterized by "paradoxical" myotonia. This type of myotonia has been termed paradoxical because it becomes worse with exercise whereas classical myotonia, as seen in myotonia congenita, is alleviated by exercise. PC is also distinguished as it can be induced by cold temperatures. Although more typical of the periodic paralytic disorders, patients with PC may also have potassium-provoked paralysis. PC typically presents within the first decade of life and has 100% penetrance. Patients with this disorder commonly present with myotonia in the face or upper extremities. The lower extremities are generally less affected. While some other related disorders result in muscle atrophy, this is not normally the case with PC. This disease can also present as hyperkalemic periodic paralysis and there is debate as to whether the two disorders are actually distinct.
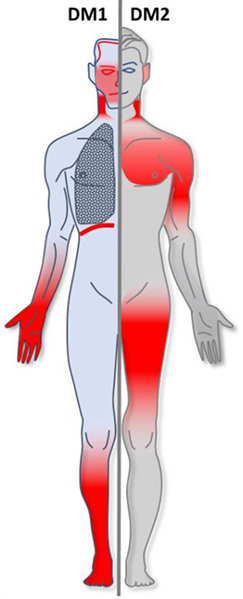
Myotonic dystrophy (DM) is a type of muscular dystrophy, a group of genetic disorders that cause progressive muscle loss and weakness. In DM, muscles are often unable to relax after contraction. Other manifestations may include cataracts, intellectual disability and heart conduction problems. In men, there may be early balding and infertility. While myotonic dystrophy can occur at any age, onset is typically in the 20s and 30s.
Congenital myopathy is a very broad term for any muscle disorder present at birth. This defect primarily affects skeletal muscle fibres and causes muscular weakness and/or hypotonia. Congenital myopathies account for one of the top neuromuscular disorders in the world today, comprising approximately 6 in 100,000 live births every year. As a whole, congenital myopathies can be broadly classified as follows:
Sarcoplasmic/endoplasmic reticulum calcium ATPase 1 (SERCA1) also known as Calcium pump 1, is an enzyme that in humans is encoded by the ATP2A1 gene.

Ryanodine receptor 1 (RYR-1) also known as skeletal muscle calcium release channel or skeletal muscle-type ryanodine receptor is one of a class of ryanodine receptors and a protein found primarily in skeletal muscle. In humans, it is encoded by the RYR1 gene.
Metabolic myopathies are myopathies that result from defects in biochemical metabolism that primarily affect muscle. They are generally genetic defects that interfere with the ability to create energy, causing a low ATP reservoir within the muscle cell.
Acquired non-inflammatory myopathy (ANIM) is a neuromuscular disorder primarily affecting skeletal muscle, most commonly in the limbs of humans, resulting in a weakness or dysfunction in the muscle. A myopathy refers to a problem or abnormality with the myofibrils, which compose muscle tissue. In general, non-inflammatory myopathies are a grouping of muscular diseases not induced by an autoimmune-mediated inflammatory pathway. These muscular diseases usually arise from a pathology within the muscle tissue itself rather than the nerves innervating that tissue. ANIM has a wide spectrum of causes which include drugs and toxins, nutritional imbalances, acquired metabolic dysfunctions such as an acquired defect in protein structure, and infections.

Schwartz–Jampel syndrome is a rare genetic disease caused by a mutation in the perlecan gene (HSPG2) which causes osteochondrodysplasia associated with myotonia. Most people with Schwartz–Jampel syndrome have a nearly normal life expectancy.
